
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date▲ | |
|---|---|---|---|---|---|---|---|---|---|
14 Nov 2022
Ehrlichia ruminantium uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cellsAdhesion process of Ehrlichia ruminantium to its host cell: the role of the protein ERGACDS01230 elucidatedRecommended by Thomas Pollet based on reviews by Rodolfo García-Contreras and Alejandro Cabezas-CruzAs recently reported by the world organisation for animal health, 60% of infectious diseases are zoonotic with a significant part associated to ticks. Ticks can transmit various pathogens such as bacteria, viruses and parasites. Among pathogens known to be transmitted by ticks, Ehrlichia ruminantium is an obligate intracellular Gram-negative bacterium responsible for the fatal heartwater disease of domestic and wild ruminants (Allsopp, 2010). E. ruminantium is transmitted by ticks of the genus Amblyomma in the tropical and sub-Saharan areas, as well as in the Caribbean islands. It constitutes a major threat for the American livestock industries since a suitable tick vector is already present in the American mainland and potential introduction of infected A. variegatum through migratory birds or uncontrolled movement of animals from Caribbean could occur (i.e. Deem, 1998 ; Kasari et al 2010). The disease is also a major obstacle to the introduction of animals from heartwater-free to heartwater-infected areas into sub-Saharan Africa and thus restrains breeding programs aiming at upgrading local stocks (Allsopp, 2010). In this context, it is essential to develop control strategies against heartwater, as developing effective vaccines, for instance. Such an objective requires a better understanding of the early interaction of E. ruminantium and its host cells and of the mechanisms associated with bacterial adhesion to the host-cell. In this study, the authors. studied the role of E. ruminantium membrane protein ERGA_CDS_01230 in the adhesion process to host bovine aortic endothelial cells (BAEC). After successfully producing the recombinant version of the protein, Pinarello et al (2022) followed the in vitro culture of E. ruminantium in BAEC and observed that the expression of the protein peaked at the extracellular infectious elementary body stages. This result would suggest the likely involvement of the protein in the early interaction of E. ruminantium with its host cells. The authors then showed using flow cytometry, and scanning electron microscopy, that beads coated with the recombinant protein adhered to BAEC. In addition, they also observed that the adhesion protein of E. ruminantium interacted with proteins of the cell's lysate, membrane and organelle fractions. Additionally, enzymatic treatment, degrading dermatan and chondroitin sulfates on the surface of BAEC, was associated with a 50% reduction in the number of bacteria in the host cell after a development cycle, indicating that glycosaminoglycans might play a role in the adhesion of E. ruminantium to the host-cell. Finally, the authors observed that the adhesion protein of E. ruminantium induced a humoral response in vaccinated animals, making this protein a possible vaccine candidate. As rightly pointed out by both reviewers, the results of this study represent a significant advance (i) in the understanding of the role of the E. ruminantium membrane protein ERGA_CDS_01230 in the adhesion process to the host-cell and (ii) in the development of new control strategies against heartwater as this protein might potentially be used as an immunogen for the development of future vaccines. References Allsopp, B.A. (2010). Natural history of Ehrlichia ruminantium. Vet Parasitol 167, 123-135. https://doi.org/10.1016/j.vetpar.2009.09.014 Deem, S.L. (1998). A review of heartwater and the threat of introduction of Cowdria ruminantium and Amblyomma spp. ticks to the American mainland. J Zoo Wildl Med 29, 109-113. Kasari, T.R. et al (2010). Recognition of the threat of Ehrlichia ruminantium infection in domestic and wild ruminants in the continental United States. J Am Vet Med Assoc. 237:520-30. https://doi.org/10.2460/javma.237.5.520 Pinarello V, Bencurova E, Marcelino I, Gros O, Puech C, Bhide M, Vachiery N, Meyer DF (2022) Ehrlichia ruminantium uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cells. bioRxiv, 2021.06.15.447525, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2021.06.15.447525 | *Ehrlichia ruminantium* uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cells | Valérie Pinarello, Elena Bencurova, Isabel Marcelino, Olivier Gros, Carinne Puech, Mangesh Bhide, Nathalie Vachiery, Damien F. Meyer | <p><em>Ehrlichia ruminantium</em> is an obligate intracellular bacterium, transmitted by ticks of the genus <em>Amblyomma</em> and responsible for heartwater, a disease of domestic and wild ruminants. High genetic diversity of <em>E. ruminantium</... | | Interactions between hosts and infectious agents/vectors, Microbiology of infections | Thomas Pollet | Rodolfo García-Contreras, Alejandro Cabezas-Cruz | 2021-10-14 16:54:54 | |
21 Jul 2022

Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV)Understanding the in vitro evolution of Cyprinid herpesvirus 3 (CyHV-3), a story of structural variations that can lead to the design of attenuated virus vaccinesRecommended by Jorge Amich based on reviews by Lucie Cappuccio and Veronique Hourdel based on reviews by Lucie Cappuccio and Veronique Hourdel
Structural variations (SVs) play a key role in viral evolution, and therefore they are also important for infection dynamics. However, the contribution of structural variations to the evolution of double-stranded viruses is limited. This knowledge can help to understand the population dynamics and might be crucial for the future development of viral attenuated vaccines. In this study, Fuandila et al (1) use the Cyprinid herpesvirus 3 (CyHV-3), commonly known as koi herpesvirus (KHV), to investigate the variability and contribution of structural variations (SV) for viral evolution after 99 passages in vitro. This virus, with the largest genome among herperviruses, causes a lethal infection in common carp and koi associated with mortalities up to 95% (2). Interestingly, KHV infections are caused by haplotype mixtures, which possibly are a source of genome diversification, but make genomic comparisons more difficult. The authors have used ultra-deep long-read sequencing of two passages, P78 and P99, which were previously described to have differences in virulence. They have found a surprisingly high and wide distribution of SVs along the genome, which were enriched in inversion and deletion events and that often led to defective viral genomes. Although it is known that these defective viral genomes negatively impact viral replication, their implications for virus persistence are still unclear. Subsequently, the authors concentrated on the virulence-relevant region ORF150, which was found to be different in P78 (deletion in 100% of the reads) and P99 (reference-like haplotype). To understand this loss and gain of full ORF150, they searched for SV turn-over in 10 intermediate passages. This analysis revealed that by passage 10 deleted and inverted (attenuated) haplotypes had already appeared, steadily increased frequency until P78, and then completely disappeared between P78 and P99. This is a striking result that raises new questions as to how this clearance occurs, which is really important as these reversions may result in undesirable increases in virulence of live-attenuated vaccines. We recommend this preprint because its use of ultra-deep long-read sequencing has permitted to better understand the role of SV diversity and dynamics in viral evolution. This study shows an unexpectedly high number of structural variations, revealing a novel source of virus diversification and confirming the different mixtures of haplotypes in different passages, including the gain of function. This research provides basic knowledge for the future design of live-attenuated vaccines, to prevent the reversion to virulent viruses. References (1) Fuandila NN, Gosselin-Grenet A-S, Tilak M-K, Bergmann SM, Escoubas J-M, Klafack S, Lusiastuti AM, Yuhana M, Fiston-Lavier A-S, Avarre J-C, Cherif E (2022) Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV). bioRxiv, 2022.03.10.483410, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.03.10.483410 (2) Sunarto A, McColl KA, Crane MStJ, Sumiati T, Hyatt AD, Barnes AC, Walker PJ. Isolation and characterization of koi herpesvirus (KHV) from Indonesia: identification of a new genetic lineage. Journal of Fish Diseases, 34, 87-101. https://doi.org/10.1111/j.1365-2761.2010.01216.x | Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV) | Nurul Novelia Fuandila, Anne-Sophie Gosselin-Grenet, Marie-Ka Tilak, Sven M Bergmann, Jean-Michel Escoubas, Sandro Klafack, Angela Mariana Lusiastuti, Munti Yuhana, Anna-Sophie Fiston-Lavier, Jean-Christophe Avarre, Emira Cherif | <p style="text-align: justify;">Structural variations (SVs) constitute a significant source of genetic variability in virus genomes. Yet knowledge about SV variability and contribution to the evolutionary process in large double-stranded (ds)DNA v... | | Animal diseases, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Viruses | Jorge Amich | Lucie Cappuccio, Veronique Hourdel | 2022-03-11 10:50:50 | |
08 Dec 2022
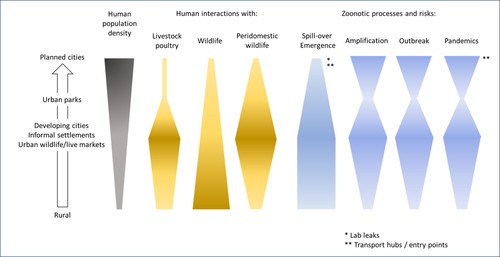
Zoonotic emergence at the animal-environment-human interface: the forgotten urban socio-ecosystemsZoonotic emergence and the overlooked case of citiesRecommended by Etienne Waleckx based on reviews by Eric Dumonteil, Nicole L. Gottdenker and 1 anonymous reviewerZoonotic pathogens, those transmitted from animals to humans, constitute a major public health risk with high associated global economic costs. Diseases associated with these pathogens represent more than 60% of emerging infectious diseases and predominantly originate in wildlife (1). Over the last decades, the emergence and re-emergence of zoonotic pathogens have led to an increasing number of epidemics, as illustrated by the current Covid-19 pandemic. There is ample evidence that human impact on native ecosystems such as deforestation, agricultural development, and urbanization, is linked to spillover of pathogens from animals to humans (2). However, research and calls to action have mainly focused on the importance of surveillance and prevention of zoonotic emergences along landscape interfaces, with special emphasis on tropical forests and agroecosystems, and studies and reviews pointing out the zoonotic risk associated with cities are scarce. Additionally, cities are sometimes wrongly seen as one homogeneous ecosystem, almost exclusively human, with a Northern hemisphere-biased perception of what a city is, which fails to take into account the ecological and socio-economic diversities that can constitute an urban area. Here, Dobigny and Morand (3) aim to draw attention to the importance of urban ecosystems in zoonotic risk and advocate that further attention should be paid to urban, peri-urban and suburban areas. In this well-organized and well-documented review, the authors show, using updated literature, that cities are places where massive contacts occur between wildlife, domestic animals, and human inhabitants (thus constituting spillover opportunities), and that it is even likely that human and wildlife contact in urban centers is more prevalent than in wild areas, perhaps contrary to intuition. Indeed, cities currently constitute the most important environment of human life and are places for millions of close interactions between humans and animals, including pets and domestic animals, wild animals through the intrusion of wild urban-adapted species (e.g., some bat, rodent, or bird species among others), manipulation and consumption of wildlife meat, and the existence of wildlife meat markets, which all constitute a major risk for zoonotic spillover. In cities, lab escapees of zoonotic pathogens also exist, and trends of adaptation to urban ecological conditions of many vectors of primary health importance is also a concern. The authors further argue that cities are predominant places for both epidemic amplification of human-human transmitted pathogens, because they are places with high human densities and population growth, and for dissemination of reservoirs, vectors and pathogens, as they are transport hubs. Dobigny & Morand further predict, likely correctly, that cities may be important places for pathogen evolution. Finally, they propose actions and recommendations to limit the risk of zoonotic spillover events from urban ecosystems and future directions for research aiming at assessing this risk. The reviewers found the manuscript well-organized and presented, timely, and bringing novel contributions to the field of zoonotic emergence. I strongly recommend this article, which should benefit a large audience, particularly in the context of the current Covid-19 pandemics and the ongoing One Health initiatives aiming at preventing future zoonotic disease emergence (4). References (1) Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P (2008) Global trends in emerging infectious diseases. Nature, 451, 990–993. https://doi.org/10.1038/nature06536 (2) White RJ, Razgour O (2020) Emerging zoonotic diseases originating in mammals: a systematic review of effects of anthropogenic land-use change. Mammal Review, 50, 336–352. https://doi.org/10.1111/mam.12201 (3) Dobigny G, Morand S (2022) Zoonotic emergence at the animal-environment-human interface: the forgotten urban socio-ecosystems. Zenodo, 6444776, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.6444776 (4) Morand S, Lajaunie C (2021) Biodiversity and COVID-19: A report and a long road ahead to avoid another pandemic. One Earth, 4, 920–923. https://doi.org/10.1016/j.oneear.2021.06.007 | Zoonotic emergence at the animal-environment-human interface: the forgotten urban socio-ecosystems | Dobigny, G. & Morand, S. | <p style="text-align: justify;">Zoonotic emergence requires spillover from animals to humans, hence animal-human interactions. A lot has been emphasized on human intrusion into wild habitats (e.g., deforestation, hunting) and the development of ag... | | Disease Ecology/Evolution, Ecohealth, Ecology of hosts, infectious agents, or vectors, Evolution of hosts, infectious agents, or vectors, One Health, Zoonoses | Etienne Waleckx | 2022-04-11 11:39:11 | ||
28 Oct 2022

Development of nine microsatellite loci for Trypanosoma lewisi, a potential human pathogen in Western Africa and South-East Asia, and preliminary population genetics analysesPreliminary population genetic analysis of Trypanosoma lewisiRecommended by Annette MacLeod based on reviews by Gabriele Schönian and 1 anonymous reviewerTrypanosoma lewisi is an atypical trypanosome species. Transmitted by fleas, it has a high prevalence and worldwide distribution in small mammals, especially rats [1]. Although not typically thought to infect humans, there has been a number of reports of human infections by T. lewisi in Asia including a case of a fatal infection in an infant [2]. The fact that the parasite is resistant to lysis by normal human serum [3] suggests that many people, especially immunocompromised individuals, may be at risk from zoonotic infections by this pathogen, particularly in regions where there is close contact with T. lewisi-infected rat fleas. Indeed, it is also possible that cryptic T. lewisi infections exist but have hitherto gone undetected. Such asymptomatic infections have been detected for a number of parasitic infections including the related parasite T. b. gambiense [4]. References
[2] Truc P, Büscher P, Cuny G, Gonzatti MI, Jannin J, Joshi P, Juyal P, Lun Z-R, Mattioli R, Pays E, Simarro PP, Teixeira MMG, Touratier L, Vincendeau P, Desquesnes M (2013) Atypical Human Infections by Animal Trypanosomes. PLOS Neglected Tropical Diseases, 7, e2256. https://doi.org/10.1371/journal.pntd.0002256 [3] Lun Z-R, Wen Y-Z, Uzureau P, Lecordier L, Lai D-H, Lan Y-G, Desquesnes M, Geng G-Q, Yang T-B, Zhou W-L, Jannin JG, Simarro PP, Truc P, Vincendeau P, Pays E (2015) Resistance to normal human serum reveals Trypanosoma lewisi as an underestimated human pathogen. Molecular and Biochemical Parasitology, 199, 58–61. https://doi.org/10.1016/j.molbiopara.2015.03.007 [4] Büscher P, Bart J-M, Boelaert M, Bucheton B, Cecchi G, Chitnis N, Courtin D, Figueiredo LM, Franco J-R, Grébaut P, Hasker E, Ilboudo H, Jamonneau V, Koffi M, Lejon V, MacLeod A, Masumu J, Matovu E, Mattioli R, Noyes H, Picado A, Rock KS, Rotureau B, Simo G, Thévenon S, Trindade S, Truc P, Reet NV (2018) Do Cryptic Reservoirs Threaten Gambiense-Sleeping Sickness Elimination? Trends in Parasitology, 34, 197–207. https://doi.org/10.1016/j.pt.2017.11.008 [5] Ségard A, Roméro A, Ravel S, Truc P, Gauthier D, Gauthier P, Dossou H-J, Sylvestre B, Houéménou G, Morand S, Chaisiri K, Noûs C, De Meeûs T (2022) Development of nine microsatellite loci for Trypanosoma lewisi, a potential human pathogen in Western Africa and South-East Asia, and preliminary population genetics analyses. Zenodo, 6460010, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.6460010 | Development of nine microsatellite loci for Trypanosoma lewisi, a potential human pathogen in Western Africa and South-East Asia, and preliminary population genetics analyses | Adeline Ségard, Audrey Romero, Sophie Ravel, Philippe Truc, Gauthier Dobigny, Philippe Gauthier, Jonas Etougbetche, Henri-Joel Dossou, Sylvestre Badou, Gualbert Houéménou, Serge Morand, Kittipong Chaisiri, Camille Noûs, Thierry deMeeûs | <p><em>Trypanosoma lewisi</em> belongs to the so-called atypical trypanosomes that occasionally affect humans. It shares the same hosts and flea vector of other medically relevant pathogenic agents as Yersinia pestis, the agent of plague. Increasi... | | Animal diseases, Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Eukaryotic pathogens/symbionts, Evolution of hosts, infectious agents, or vectors, Microbiology of infections, Parasites, Population genetics of hosts, in... | Annette MacLeod | 2022-04-21 17:04:37 | ||
07 Feb 2023

Three-way relationships between gut microbiota, helminth assemblages and bacterial infections in wild rodent populationsUnveiling the complex interactions between members of gut microbiomes: a significant advance provided by an exhaustive study of wild bank volesRecommended by Thomas Pollet based on reviews by Jason Anders and 1 anonymous reviewerThe gut of vertebrates is a host for hundreds or thousands of different species of microorganisms named the gut microbiome. This latter may differ greatly in natural environments between individuals, populations and species (1). The vertebrate gut microbiome plays key roles in host fitness through functions including nutrient acquisition, immunity and defense against infectious agents. While bank voles are small mammals potentially reservoirs of a large number of infectious agents, questions about the links between their gut microbiome and the presence of pathogens are scarcely addressed. In this study, Bouilloud et al. (2) used complementary analyses of community and microbial ecology to (i) assess the variability of gut bacteriome diversity and composition in wild populations of the bank vole Myodes glareolus collected in four different sites in Eastern France and (ii) evaluate the three-way interactions between the gut bacteriota, the gastro-intestinal helminths and pathogenic bacteria detected in the spleen. Authors identified important variations of the gut bacteriota composition and diversity among bank voles mainly explained by sampling localities. They found positive correlations between the specific richness of both the gut bacteria and the helminth community, as well as between the composition of these two communities, even when accounting for the influence of geographical distance. The helminths Aonchotheca murissylvatici, Heligmosomum mixtum and the bacteria Bartonella sp were the main taxa associated with the whole gut bacteria composition. Besides, changes in relative abundance of particular gut bacterial taxa were specifically associated with other helminths (Mastophorus muris, Catenotaenia henttoneni, Paranoplocephala omphalodes and Trichuris arvicolae) or pathogenic bacteria. Infections with Neoehrlichia mikurensis, Orientia sp, Rickettsia sp and P. omphalodes were especially associated with lower relative abundance of members of the family Erysipelotrichaceae (Firmicutes), while coinfections with higher number of bacterial infections were associated with lower relative abundance of members of the Bacteroidales family (Bacteroidetes). As pointed out by both reviewers, this study represents a significant advance in the field. I would like to commend the authors for this enormous work. The amount of data, analyses and results is considerable which has sometimes complicated the understanding of the story at the beginning of the evaluation process. Thanks to constructive scientific interactions with both reviewers through the two rounds of evaluation, the authors have efficiently addressed the reviewer's concerns and improved the manuscript, making this great story easier to read. The innovative results of this study emphasize the complex interlinkages between gut bacteriome and infections in wild animal populations and I strongly recommend this article for publication In Peer Community Infections. References (1) Vujkovic-Cvijin I, Sklar J, Jiang L, Natarajan L, Knight R, Belkaid Y (2020) Host variables confound gut microbiota studies of human disease. Nature, 587, 448–454. https://doi.org/10.1038/s41586-020-2881-9 (2) Bouilloud M, Galan M, Dubois A, Diagne C, Marianneau P, Roche B, Charbonnel N (2023) Three-way relationships between gut microbiota, helminth assemblages and bacterial infections in wild rodent populations. biorxiv, 2022.05.23.493084, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.05.23.493084 | Three-way relationships between gut microbiota, helminth assemblages and bacterial infections in wild rodent populations | Marie Bouilloud, Maxime Galan, Adelaide Dubois, Christophe Diagne, Philippe Marianneau, Benjamin Roche, Nathalie Charbonnel | <p>Background</p> <p>Despite its central role in host fitness, the gut microbiota may differ greatly between individuals. This variability is often mediated by environmental or host factors such as diet, genetics, and infections. Recently, a part... | | Disease Ecology/Evolution, Ecohealth, Interactions between hosts and infectious agents/vectors, Reservoirs, Zoonoses | Thomas Pollet | 2022-05-25 10:13:23 | ||
07 Oct 2022

Guidelines for the reliable use of high throughput sequencing technologies to detect plant pathogens and pestsHigh-throughput sequencing for the diagnostic of plant pathologies and identification of pests: recommendations and challengesRecommended by Olivier Schumpp based on reviews by Denise Altenbach and David RoquisHigh-throughput sequencing (HTS) has revealed an incredible diversity of microorganisms in ecosystems and is also changing the monitoring of macroorganism biodiversity (Deiner et al. 2017; Piper et al. 2019). The diagnostic of plant pathogens and the identification of pests is gradually integrating the use of these techniques, but there are still obstacles. Most of them are related to the reliability of these analyses, which have long been considered insufficient because of their dependence on a succession of sophisticated operations involving parameters that are sometimes difficult to adapt to complex matrices or certain diagnostic contexts. The need to validate HTS approaches is gradually being highlighted in recent work but remains poorly documented (Bester et al. 2022). In this paper, a large community of experts presents and discusses the key steps for optimal control of HTS performance and reliability in a diagnostic context (Massart et al. 2022). It also addresses the issue of costs. The article provides recommendations that closely combine the quality control requirements commonly used in conventional diagnostics with newer or HTS-specific control elements and concepts that are not yet widely used. It discusses the value of these for the use of the various techniques currently covered by the terms "High Throughput Sequencing" in diagnostic activities. The elements presented are intended to limit false positive or false negative results but will also optimise the interpretation of contentious results close to the limits of analytical sensitivity or unexpected results, both of which appear to be frequent when using HTS. Furthermore, the need for risk analysis, verification and validation of methods is well illustrated with numerous examples for each of the steps considered crucial to ensure reliable use of HTS. The clear contextualisation of the proposals made by the authors complements and clarifies the need for user expertise according to the experimental objectives. Some unanswered questions that will require further development and validation are also presented. This article should benefit a large audience including researchers with some level of expertise in HTS but unfamiliar with the recent concepts of controls common in the diagnostic world as well as scientists with strong diagnostic expertise but less at ease with the numerous and complex procedures associated with HTS. References Bester R, Steyn C, Breytenbach JHJ, de Bruyn R, Cook G, Maree HJ (2022) Reproducibility and Sensitivity of High-Throughput Sequencing (HTS)-Based Detection of Citrus Tristeza Virus and Three Citrus Viroids. Plants, 11, 1939. https://doi.org/10.3390/plants11151939 Deiner K, Bik HM, Mächler E, Seymour M, Lacoursière-Roussel A, Altermatt F, Creer S, Bista I, Lodge DM, de Vere N, Pfrender ME, Bernatchez L (2017) Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Molecular Ecology, 26, 5872–5895. https://doi.org/10.1111/mec.14350 Massart, S et al. (2022) Guidelines for the reliable use of high throughput sequencing technologies to detect plant pathogens and pests. Zenodo, 6637519, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.6637519 Piper AM, Batovska J, Cogan NOI, Weiss J, Cunningham JP, Rodoni BC, Blacket MJ (2019) Prospects and challenges of implementing DNA metabarcoding for high-throughput insect surveillance. GigaScience, 8, giz092. https://doi.org/10.1093/gigascience/giz092 | Guidelines for the reliable use of high throughput sequencing technologies to detect plant pathogens and pests | S. Massart, I. Adams, M. Al Rwahnih, S. Baeyen, G. J. Bilodeau, A. G. Blouin, N. Boonham, T. Candresse, A. Chandelier, K. De Jonghe, A. Fox, Y.Z.A. Gaafar, P. Gentit, A. Haegeman, W. Ho, O. Hurtado-Gonzales, W. Jonkers, J. Kreuze, D. Kutjnak, B. B... | <p style="text-align: justify;">High-throughput sequencing (HTS) technologies have the potential to become one of the most significant advances in molecular diagnostics. Their use by researchers to detect and characterize plant pathogens and pests... | | Diagnosis, Pest management, Phytopathology, Plant diseases | Olivier Schumpp | 2022-06-13 11:26:18 | ||
23 Jan 2023

Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolenseWhole genome transcriptome reveals metabolic and immune susceptibility factors for Trypanosoma congolense infection in West-African livestockRecommended by Concepción Marañón based on reviews by 2 anonymous reviewersAfrican trypanosomiasis is caused by to the infection of a protozoan parasite of the Trypanosoma genus. It is transmitted by the tsetse fly, and is largely affecting cattle in the sub-humid areas of Africa, causing a high economic impact. However, not all the bovine strains are equally susceptible to the infection (1). In order to dissect the mechanisms underlying susceptibility to African trypanosoma infection, Peylhard et al (2) performed blood transcriptional profiles of trypanotolerant, trypanosensitive and mixed cattle breeds, before and after experimental infection with T. congolense. First of all, the authors have characterized the basal transcriptional profiles in the blood of the different breeds under study, which could be classified in a wide array of functional pathways. Of note, after infection some pathways were consistently enriched in all the group tested. Among them, the immune system-related ones were again on the top functions reported. The search for specific canonical pathways pointed to a prominent role of lipid and cholesterol-related pathways, as well as mitochondrial function and B and T lymphocyte activation. However, the analysis of infected animals demonstrated that trypanosusceptible animals showed a stronger transcriptomic reprogramming, highly enriched in specific metabolic and immunological pathways. It is worthy to highlight striking differences in genes involved in immune signal transduction, cytokines and markers of different leukocyte subpopulations. This work represents undoubtedly a significant momentum in the field, since the authors explore in deep a wide panel of cattle breeds representing the majority of West-African taurine and zebu in a systematic way. Since the animals were studied at different timepoints after infection, future longitudinal analyses of these datasets will be providing a precious insight on the kinetics of immune and metabolic reprogramming associated with susceptibility and tolerance to African trypanosoma infection, widening the application of this interesting study into new therapeutic interventions. References 1. Berthier D, Peylhard M, Dayo G-K, Flori L, Sylla S, Bolly S, Sakande H, Chantal I, Thevenon S (2015) A Comparison of Phenotypic Traits Related to Trypanotolerance in Five West African Cattle Breeds Highlights the Value of Shorthorn Taurine Breeds. PLOS ONE, 10, e0126498. https://doi.org/10.1371/journal.pone.0126498 2. Peylhard M, Berthier D, Dayo G-K, Chantal I, Sylla S, Nidelet S, Dubois E, Martin G, Sempéré G, Flori L, Thévenon S (2022) Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense. bioRxiv, 2022.06.10.495622, ver. 2 peer-reviewed and recommended by Peer Community Infections. https://doi.org/10.1101/2022.06.10.495622. | Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense | Moana Peylhard, David Berthier, Guiguigbaza-Kossigan Dayo, Isabelle Chantal, Souleymane Sylla, Sabine Nidelet, Emeric Dubois, Guillaume Martin, Guilhem Sempéré, Laurence Flori, Sophie Thévenon | <p>Animal African trypanosomosis, caused by blood protozoan parasites transmitted mainly by tsetse flies, represents a major constraint for millions of cattle in sub-Saharan Africa. Exposed cattle include trypanosusceptible indicine breeds, severe... | | Animal diseases, Genomics, functional genomics of hosts, infectious agents, or vectors, Resistance/Virulence/Tolerance | Concepción Marañón | Anonymous, Anonymous | 2022-06-14 17:06:57 | |
14 Dec 2022

Transcriptome responses of the aphid vector Myzus persicae are shaped by identities of the host plant and the virusHow do multiple host plants and virus species challenge aphid molecular machinery?Recommended by Sebastien Massart based on reviews by Juan José Lopez Moya and Michelle HeckThe impact of virus infection of a plant on an aphid’s behaviour has been observed in many studies [1]. Indeed, virus infection can alter plant biochemistry through the emission of volatile organic compounds and plant tissue content modification. These alterations can further impact the interactions between plants and aphids. However, although it is a well-known phenomenon, very few studies have explored the consequences of plant virus infection on the gene expression of aphids to understand better the aphid’s manipulation by the plant virus. In this context, the recommended study [2] reports a comprehensive transcriptomic analysis of the genes expressed by one aphid species, Myzus persicae, a vector of several plant viruses, when feeding on plants. Michelle Heck underlined how significant this study is for comprehending the molecular bases of aphid-vector manipulation by plant viruses (see below). Interestingly, the study design has integrated several factors that might influence the gene expression of M. persicae when feeding on the plant. Indeed, the authors investigated the effect of two plant species (Arabidopsis thaliana and Camelia sativa) and two virus species [turnip yellows virus (TuYV) and cauliflower mosaic virus (CaMV)]. Noteworthy, the transmission mode of TuYV is circulative and persistent, while CaMV is transmitted by a semi-persistent non-circulative mode. As Juan José Lopez Moya mentioned, multiple comparisons allowed the identification of the different responses of aphids in front of different host plants infected or not by different viruses (see below). This publication is complementary to a previous publication from the same team focusing on plant transcriptome analysis [3]. Thanks to their experimental design, the authors identified genes commonly deregulated by both viruses and/or both plant species and deregulated genes by a single virus or a single plant. Figure 4 nicely summarizes the number of deregulated genes. A thorough discussion on the putative role of deregulated genes in different conditions gave a comprehensive follow-up of the results and their impact on the current knowledge of plant-virus-vector interactions. This study has now opened the gate to promising research focusing on the functional validation of the identified genes while also narrowing the study from the body to the tissue level. References: 1. Carr JP, Tungadi T, Donnelly R, Bravo-Cazar A, Rhee S-J, Watt LG, Mutuku JM, Wamonje FO, Murphy AM, Arinaitwe W, Pate AE, Cunniffe NJ, Gilligan CA (2020) Modelling and manipulation of aphid-mediated spread of non-persistently transmitted viruses. Virus Research, 277, 197845. https://doi.org/10.1016/j.virusres.2019.197845 2. Chesnais Q, Golyaev V, Velt A, Rustenholz C, Verdier M, Brault V, Pooggin MM, Drucker M (2022) Transcriptome responses of the aphid vector Myzus persicae are shaped by identities of the host plant and the virus. bioRxiv , 2022.07.18.500449, ver. 5 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.07.18.500449 3. Chesnais Q, Golyaev V, Velt A, Rustenholz C, Brault V, Pooggin MM, Drucker M (2022) Comparative Plant Transcriptome Profiling of Arabidopsis thaliana Col-0 and Camelina sativa var. Celine Infested with Myzus persicae Aphids Acquiring Circulative and Noncirculative Viruses Reveals Virus- and Plant-Specific Alterations Relevant to Aphid Feeding Behavior and Transmission. Microbiology Spectrum, 10, e00136-22. https://doi.org/10.1128/spectrum.00136-22 | Transcriptome responses of the aphid vector *Myzus persicae* are shaped by identities of the host plant and the virus | Quentin Chesnais, Victor Golyaev, Amandine Velt, Camille Rustenholz, Maxime Verdier, Véronique Brault, Mikhail M. Pooggin, Martin Drucker | <p style="text-align: justify;"><strong>Background:</strong> Numerous studies have documented modifications in vector orientation behavior, settling and feeding behavior, and/or fecundity and survival due to virus infection in host plants. These a... | | Behaviour of hosts, infectious agents, or vectors, Cell biology of hosts, infectious agents, or vectors, Molecular biology of infections, Physiology of hosts, infectious agents, or vectors, Phytopathology, Plant diseases, Vectors, Viruses | Sebastien Massart | 2022-07-19 15:24:14 | ||
06 Apr 2023

Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populationsChanges in aggressiveness in pathotypes of wheat leaf rustRecommended by Pierre Gladieux based on reviews by 2 anonymous reviewersUnderstanding the ecological and evolutionary factors underlying the spread of new fungal pathogen populations can inform the development of more effective management strategies. In plant pathology, pathogenicity is generally presented as having two components: ‘virulence’ (qualitative pathogenicity) and aggressiveness (quantitative pathogenicity). Changes in virulence in response to the deployment of new resistant varieties are a major driver of the spread of new populations (called pathotypes, or races) in modern agrosystems, and the genomic (i.e. proximal) and eco-evolutionary (i.e. ultimate) factors underlying these changes are well-documented [1,2,3]. By contrast, the role of changes in aggressiveness in the spread of pathotypes remains little known [4]. The study by Cécilia Fontyn and collaborators [5] set out to characterize changes in aggressiveness for isolates of two pathotypes of the wheat leaf rust (Puccinia triticina) that have been dominant in France during the 2005-2016 period. Isolates were genetically characterized using multilocus microsatellite typing and phenotypically characterized for three components of aggressiveness on wheat varieties: infection efficiency, latency period, and sporulation capacity. Using experiments that represent quite a remarkable amount of work and effort, Fontyn et al. showed that each dominant pathotype consisted of several genotypes, including common genotypes whose frequency changed over time. For each pathotype, the genotypes that were more common initially were replaced by a more aggressive genotype. Together, these results show that changes in the genetic composition of populations of fungal plant pathogens can be associated with, and may be caused by, changes in the quantitative components of pathogenicity. This study also illustrates how extensive, decade-long monitoring of fungal pathogen populations, such as the one conducted for wheat leaf rust in France, represents a very valuable resource for research. REFERENCES [1] Brown, J. K. (1994). Chance and selection in the evolution of barley mildew. Trends in Microbiology, 2(12), 470-475. https://doi.org/10.1016/0966-842x(94)90650-5 [2] Daverdin, G., Rouxel, T., Gout, L., Aubertot, J. N., Fudal, I., Meyer, M., Parlange, F., Carpezat, J., & Balesdent, M. H. (2012). Genome structure and reproductive behaviour influence the evolutionary potential of a fungal phytopathogen. PLoS Pathogens, 8(11), e1003020. https://doi.org/10.1371/journal.ppat.1003020 [3] Gladieux, P., Feurtey, A., Hood, M. E., Snirc, A., Clavel, J., Dutech, C., Roy, M., & Giraud, T. (2015). The population biology of fungal invasions.Molecular Ecology, 24(9), 1969-86. https://doi.org/10.1111/mec.13028 [4] Fontyn, C., Zippert, A. C., Delestre, G., Marcel, T. C., Suffert, F., & Goyeau, H. (2022). Is virulence phenotype evolution driven exclusively by Lr gene deployment in French Puccinia triticina populations?. Plant Pathology, 71(7), 1511-1524. https://doi.org/10.1111/ppa.13599 [5] Fontyn, C., Meyer, K. J., Boixel, A. L., Delestre, G., Piaget, E., Picard, C., Suffer, F., Marcel, T.C., & Goyeau, H. (2022). Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations. bioRxiv, 2022.08.29.505401, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.29.505401 | Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations | Cécilia FONTYN, Kevin JG MEYER, Anne-Lise BOIXEL, Ghislain DELESTRE, Emma PIAGET, Corentin PICARD, Frédéric SUFFERT, Thierry C MARCEL, Henriette GOYEAU | <p style="text-align: justify;">Plant pathogens are constantly evolving and adapting to their environment, including their host. Virulence alleles emerge, and then increase, and sometimes decrease in frequency within pathogen populations in respon... | | Coevolution, Epidemiology, Evolution of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Pathogenic/Symbiotic Fungi, Phytopathology, Plant diseases, Population dynamics of hosts, infectious agents, or... | Pierre Gladieux | Emerson Del Ponte , Jacqui Shykoff, Leïla Bagny Beilhe , Alexey Mikaberidze | 2022-09-29 20:01:57 | |
23 Mar 2023
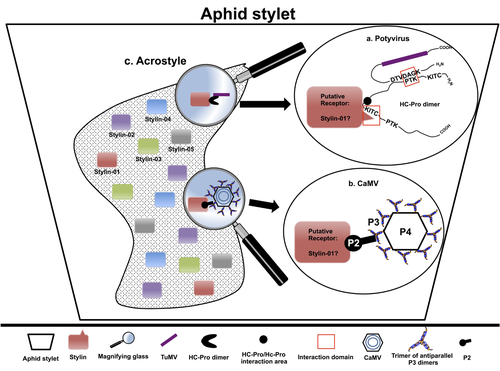
The helper strategy in vector-transmission of plant virusesThe intriguing success of helper components in vector-transmission of plant viruses.Recommended by Christine Coustau based on reviews by Jamie Bojko and Olivier SchumppMost plant-infecting viruses rely on an animal vector to be transmitted from one sessile host plant to another. A fascinating aspect of virus-vector interactions is the fact that viruses from different clades produce different proteins to bind vector receptors (1). Two major processes are described. In the “capsid strategy”, a motif of the capsid protein is directly binding to the vector receptor. In the “helper strategy”, a non-structural component, the helper component (HC), establishes a bridge between the virus particle and the vector’s receptor. In this exhaustive review focusing on hemipteran insect vectors, Di Mattia et al. (2) are revisiting the helper strategy in light of recent results. The authors first place the discoveries of the HC strategy in a historical context, suggesting that HC are exclusively found in non-circulative viruses (viruses that only attach to the vector). They present an overview of the nature and modes of action of helper components in the major virus clades of non-circulative viruses (Potyviruses and Caulimoviruses). Authors then detail recent advances, to which they have significantly contributed, showing that the helper strategy also appears widespread in circulative transmission categories (Tenuiviruses, Nanoviruses). In an extensive perspective section, they raise the question of the evolutionary significance of the existence of HC in numerous unrelated viruses, transmitted by unrelated vectors through different mechanisms. They explore the hypothesis that the helper strategy evolved several times independently in distinct viral clades and for different reasons. In particular, they present several potential benefits of plant virus HC related to virus cooperation, collective transmission and effector-driven infectivity. As pointed out by both reviewers, this is a very clear and synthetic review. Di Mattia et al. present an exhaustive overview of virus HC-vector molecular interactions and address functionally and evolutionarily important questions. This review should benefit a large audience interested in host-virus interactions and transmission processes. REFERENCES (1) Ng JCK, Falk BW (2006) Virus-Vector Interactions Mediating Nonpersistent and Semipersistent Transmission of Plant Viruses. Annual Review of Phytopathology, 44, 183–212. https://doi.org/10.1146/annurev.phyto.44.070505.143325 (2) Di Mattia J, Zeddam J-L, Uzest M, Blanc S (2023) The helper strategy in vector-transmission of plant viruses. Zenodo, ver. 2 peer-reviewed and recommended by Peer Community In Infections. https://doi.org/10.5281/zenodo.7709290 | The helper strategy in vector-transmission of plant viruses | Di Mattia Jérémy, Zeddam Jean Louis, Uzest Marilyne and Stéphane Blanc | <p>An intriguing aspect of vector-transmission of plant viruses is the frequent involvement of a helper component (HC). HCs are virus-encoded non-structural proteins produced in infected plant cells that are mandatory for the transmission success.... | | Evolution of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Molecular biology of infections, Molecular genetics of hosts, infectious agents, or vectors, Plant diseases, Vectors, Viruses | Christine Coustau | 2022-10-28 17:32:39 |




