
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * ▲ | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
08 Aug 2023
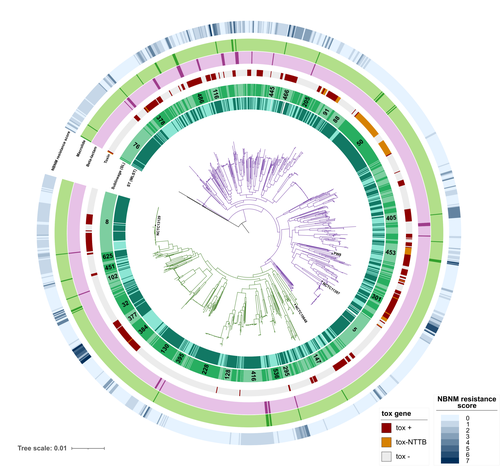
A global Corynebacterium diphtheriae genomic framework sheds light on current diphtheria reemergenceDIPHTOSCAN : A new tool for the genomic surveillance of diphtheriaRecommended by Rodolfo García-Contreras based on reviews by Ankur Mutreja and 2 anonymous reviewersOne of the greatest achievements of health sciences is the eradication of infectious diseases such as smallpox that in the past imposed a severe burden on humankind, through global vaccination campaigns. Moreover, progress towards the eradication of others such as poliomyelitis, dracunculiasis, and yaws is being made. In contrast, other infections that were previously contained are reemerging, due to several factors, including lack of access to vaccines due to geopolitical reasons, the rise of anti-vaccine movements, and the constant mobility of infected persons from the endemic sites. One of such disease is diphtheria, caused by Corynebacterium diphtheriae and a few other related species such as C. ulcerans and C. pseudotuberculosis. Importantly, in France, diphtheria cases reported in 2022 increased 7-fold from the average of previously recorded cases per year in the previous 4 years and the situation in other European countries is similar. Hence, as reported here, Hennart et al. (2023) developed DIPHTOSCAN, a free access bioinformatics tool with user-friendly interphase, aimed to easily identify, extract and interpret important genomic features such as the sublineage of the strain, the presence of the tox gene (as a string predictor for toxigenic disease) as well as genes coding other virulence factors such as fimbriae, and the presence of know resistant mechanisms towards antibiotics like penicillin and erythromycin currently used in the clinic to treat this infection. The authors validated the performance of their tool with a large collection of genomes, including those obtained from the isolates of the 2022 outbreak in France, more than 1,200 other genomes isolated from France, Algeria, and Yemen, and more than 500 genomes from several countries from Europe, America, Africa, Asia, and Oceania that are available through the NCBI site. DIPHTOSCAN will allow the rapid identification and surveillance of potentially dangerous strains such as those being tox-positive isolates and resistant to multiple drugs and/or first-line treatments and a better understanding of the epidemiology and evolution of this important reemerging disease. | A global *Corynebacterium diphtheriae* genomic framework sheds light on current diphtheria reemergence | Melanie Hennart, Chiara Crestani, Sebastien Bridel, Nathalie Armatys, Sylvie Brémont, Annick Carmi-Leroy, Annie Landier, Virginie Passet, Laure Fonteneau, Sophie Vaux, Julie Toubiana, Edgar Badell, Sylvain Brisse | <p style="text-align: justify;"><strong>Background</strong></p> <p style="text-align: justify;">Diphtheria, caused by <em>Corynebacterium diphtheriae</em>, reemerges in Europe since 2022. Genomic sequencing can inform on transmission routes and g... | | Drug resistance, tolerance and persistence, Epidemiology, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Microbiology of infections, Population genetics of hosts, infectiou... | Rodolfo García-Contreras | Ankur Mutreja | 2023-03-09 16:02:27 | |
14 Dec 2022

Transcriptome responses of the aphid vector Myzus persicae are shaped by identities of the host plant and the virusHow do multiple host plants and virus species challenge aphid molecular machinery?Recommended by Sebastien Massart based on reviews by Juan José Lopez Moya and Michelle HeckThe impact of virus infection of a plant on an aphid’s behaviour has been observed in many studies [1]. Indeed, virus infection can alter plant biochemistry through the emission of volatile organic compounds and plant tissue content modification. These alterations can further impact the interactions between plants and aphids. However, although it is a well-known phenomenon, very few studies have explored the consequences of plant virus infection on the gene expression of aphids to understand better the aphid’s manipulation by the plant virus. In this context, the recommended study [2] reports a comprehensive transcriptomic analysis of the genes expressed by one aphid species, Myzus persicae, a vector of several plant viruses, when feeding on plants. Michelle Heck underlined how significant this study is for comprehending the molecular bases of aphid-vector manipulation by plant viruses (see below). Interestingly, the study design has integrated several factors that might influence the gene expression of M. persicae when feeding on the plant. Indeed, the authors investigated the effect of two plant species (Arabidopsis thaliana and Camelia sativa) and two virus species [turnip yellows virus (TuYV) and cauliflower mosaic virus (CaMV)]. Noteworthy, the transmission mode of TuYV is circulative and persistent, while CaMV is transmitted by a semi-persistent non-circulative mode. As Juan José Lopez Moya mentioned, multiple comparisons allowed the identification of the different responses of aphids in front of different host plants infected or not by different viruses (see below). This publication is complementary to a previous publication from the same team focusing on plant transcriptome analysis [3]. Thanks to their experimental design, the authors identified genes commonly deregulated by both viruses and/or both plant species and deregulated genes by a single virus or a single plant. Figure 4 nicely summarizes the number of deregulated genes. A thorough discussion on the putative role of deregulated genes in different conditions gave a comprehensive follow-up of the results and their impact on the current knowledge of plant-virus-vector interactions. This study has now opened the gate to promising research focusing on the functional validation of the identified genes while also narrowing the study from the body to the tissue level. References: 1. Carr JP, Tungadi T, Donnelly R, Bravo-Cazar A, Rhee S-J, Watt LG, Mutuku JM, Wamonje FO, Murphy AM, Arinaitwe W, Pate AE, Cunniffe NJ, Gilligan CA (2020) Modelling and manipulation of aphid-mediated spread of non-persistently transmitted viruses. Virus Research, 277, 197845. https://doi.org/10.1016/j.virusres.2019.197845 2. Chesnais Q, Golyaev V, Velt A, Rustenholz C, Verdier M, Brault V, Pooggin MM, Drucker M (2022) Transcriptome responses of the aphid vector Myzus persicae are shaped by identities of the host plant and the virus. bioRxiv , 2022.07.18.500449, ver. 5 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.07.18.500449 3. Chesnais Q, Golyaev V, Velt A, Rustenholz C, Brault V, Pooggin MM, Drucker M (2022) Comparative Plant Transcriptome Profiling of Arabidopsis thaliana Col-0 and Camelina sativa var. Celine Infested with Myzus persicae Aphids Acquiring Circulative and Noncirculative Viruses Reveals Virus- and Plant-Specific Alterations Relevant to Aphid Feeding Behavior and Transmission. Microbiology Spectrum, 10, e00136-22. https://doi.org/10.1128/spectrum.00136-22 | Transcriptome responses of the aphid vector *Myzus persicae* are shaped by identities of the host plant and the virus | Quentin Chesnais, Victor Golyaev, Amandine Velt, Camille Rustenholz, Maxime Verdier, Véronique Brault, Mikhail M. Pooggin, Martin Drucker | <p style="text-align: justify;"><strong>Background:</strong> Numerous studies have documented modifications in vector orientation behavior, settling and feeding behavior, and/or fecundity and survival due to virus infection in host plants. These a... | | Behaviour of hosts, infectious agents, or vectors, Cell biology of hosts, infectious agents, or vectors, Molecular biology of infections, Physiology of hosts, infectious agents, or vectors, Phytopathology, Plant diseases, Vectors, Viruses | Sebastien Massart | 2022-07-19 15:24:14 | ||
14 Nov 2022
Ehrlichia ruminantium uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cellsAdhesion process of Ehrlichia ruminantium to its host cell: the role of the protein ERGACDS01230 elucidatedRecommended by Thomas Pollet based on reviews by Rodolfo García-Contreras and Alejandro Cabezas-CruzAs recently reported by the world organisation for animal health, 60% of infectious diseases are zoonotic with a significant part associated to ticks. Ticks can transmit various pathogens such as bacteria, viruses and parasites. Among pathogens known to be transmitted by ticks, Ehrlichia ruminantium is an obligate intracellular Gram-negative bacterium responsible for the fatal heartwater disease of domestic and wild ruminants (Allsopp, 2010). E. ruminantium is transmitted by ticks of the genus Amblyomma in the tropical and sub-Saharan areas, as well as in the Caribbean islands. It constitutes a major threat for the American livestock industries since a suitable tick vector is already present in the American mainland and potential introduction of infected A. variegatum through migratory birds or uncontrolled movement of animals from Caribbean could occur (i.e. Deem, 1998 ; Kasari et al 2010). The disease is also a major obstacle to the introduction of animals from heartwater-free to heartwater-infected areas into sub-Saharan Africa and thus restrains breeding programs aiming at upgrading local stocks (Allsopp, 2010). In this context, it is essential to develop control strategies against heartwater, as developing effective vaccines, for instance. Such an objective requires a better understanding of the early interaction of E. ruminantium and its host cells and of the mechanisms associated with bacterial adhesion to the host-cell. In this study, the authors. studied the role of E. ruminantium membrane protein ERGA_CDS_01230 in the adhesion process to host bovine aortic endothelial cells (BAEC). After successfully producing the recombinant version of the protein, Pinarello et al (2022) followed the in vitro culture of E. ruminantium in BAEC and observed that the expression of the protein peaked at the extracellular infectious elementary body stages. This result would suggest the likely involvement of the protein in the early interaction of E. ruminantium with its host cells. The authors then showed using flow cytometry, and scanning electron microscopy, that beads coated with the recombinant protein adhered to BAEC. In addition, they also observed that the adhesion protein of E. ruminantium interacted with proteins of the cell's lysate, membrane and organelle fractions. Additionally, enzymatic treatment, degrading dermatan and chondroitin sulfates on the surface of BAEC, was associated with a 50% reduction in the number of bacteria in the host cell after a development cycle, indicating that glycosaminoglycans might play a role in the adhesion of E. ruminantium to the host-cell. Finally, the authors observed that the adhesion protein of E. ruminantium induced a humoral response in vaccinated animals, making this protein a possible vaccine candidate. As rightly pointed out by both reviewers, the results of this study represent a significant advance (i) in the understanding of the role of the E. ruminantium membrane protein ERGA_CDS_01230 in the adhesion process to the host-cell and (ii) in the development of new control strategies against heartwater as this protein might potentially be used as an immunogen for the development of future vaccines. References Allsopp, B.A. (2010). Natural history of Ehrlichia ruminantium. Vet Parasitol 167, 123-135. https://doi.org/10.1016/j.vetpar.2009.09.014 Deem, S.L. (1998). A review of heartwater and the threat of introduction of Cowdria ruminantium and Amblyomma spp. ticks to the American mainland. J Zoo Wildl Med 29, 109-113. Kasari, T.R. et al (2010). Recognition of the threat of Ehrlichia ruminantium infection in domestic and wild ruminants in the continental United States. J Am Vet Med Assoc. 237:520-30. https://doi.org/10.2460/javma.237.5.520 Pinarello V, Bencurova E, Marcelino I, Gros O, Puech C, Bhide M, Vachiery N, Meyer DF (2022) Ehrlichia ruminantium uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cells. bioRxiv, 2021.06.15.447525, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2021.06.15.447525 | *Ehrlichia ruminantium* uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cells | Valérie Pinarello, Elena Bencurova, Isabel Marcelino, Olivier Gros, Carinne Puech, Mangesh Bhide, Nathalie Vachiery, Damien F. Meyer | <p><em>Ehrlichia ruminantium</em> is an obligate intracellular bacterium, transmitted by ticks of the genus <em>Amblyomma</em> and responsible for heartwater, a disease of domestic and wild ruminants. High genetic diversity of <em>E. ruminantium</... | | Interactions between hosts and infectious agents/vectors, Microbiology of infections | Thomas Pollet | Rodolfo García-Contreras, Alejandro Cabezas-Cruz | 2021-10-14 16:54:54 | |
08 Dec 2022
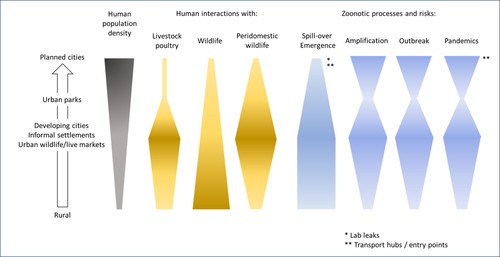
Zoonotic emergence at the animal-environment-human interface: the forgotten urban socio-ecosystemsZoonotic emergence and the overlooked case of citiesRecommended by Etienne Waleckx based on reviews by Eric Dumonteil, Nicole L. Gottdenker and 1 anonymous reviewerZoonotic pathogens, those transmitted from animals to humans, constitute a major public health risk with high associated global economic costs. Diseases associated with these pathogens represent more than 60% of emerging infectious diseases and predominantly originate in wildlife (1). Over the last decades, the emergence and re-emergence of zoonotic pathogens have led to an increasing number of epidemics, as illustrated by the current Covid-19 pandemic. There is ample evidence that human impact on native ecosystems such as deforestation, agricultural development, and urbanization, is linked to spillover of pathogens from animals to humans (2). However, research and calls to action have mainly focused on the importance of surveillance and prevention of zoonotic emergences along landscape interfaces, with special emphasis on tropical forests and agroecosystems, and studies and reviews pointing out the zoonotic risk associated with cities are scarce. Additionally, cities are sometimes wrongly seen as one homogeneous ecosystem, almost exclusively human, with a Northern hemisphere-biased perception of what a city is, which fails to take into account the ecological and socio-economic diversities that can constitute an urban area. Here, Dobigny and Morand (3) aim to draw attention to the importance of urban ecosystems in zoonotic risk and advocate that further attention should be paid to urban, peri-urban and suburban areas. In this well-organized and well-documented review, the authors show, using updated literature, that cities are places where massive contacts occur between wildlife, domestic animals, and human inhabitants (thus constituting spillover opportunities), and that it is even likely that human and wildlife contact in urban centers is more prevalent than in wild areas, perhaps contrary to intuition. Indeed, cities currently constitute the most important environment of human life and are places for millions of close interactions between humans and animals, including pets and domestic animals, wild animals through the intrusion of wild urban-adapted species (e.g., some bat, rodent, or bird species among others), manipulation and consumption of wildlife meat, and the existence of wildlife meat markets, which all constitute a major risk for zoonotic spillover. In cities, lab escapees of zoonotic pathogens also exist, and trends of adaptation to urban ecological conditions of many vectors of primary health importance is also a concern. The authors further argue that cities are predominant places for both epidemic amplification of human-human transmitted pathogens, because they are places with high human densities and population growth, and for dissemination of reservoirs, vectors and pathogens, as they are transport hubs. Dobigny & Morand further predict, likely correctly, that cities may be important places for pathogen evolution. Finally, they propose actions and recommendations to limit the risk of zoonotic spillover events from urban ecosystems and future directions for research aiming at assessing this risk. The reviewers found the manuscript well-organized and presented, timely, and bringing novel contributions to the field of zoonotic emergence. I strongly recommend this article, which should benefit a large audience, particularly in the context of the current Covid-19 pandemics and the ongoing One Health initiatives aiming at preventing future zoonotic disease emergence (4). References (1) Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P (2008) Global trends in emerging infectious diseases. Nature, 451, 990–993. https://doi.org/10.1038/nature06536 (2) White RJ, Razgour O (2020) Emerging zoonotic diseases originating in mammals: a systematic review of effects of anthropogenic land-use change. Mammal Review, 50, 336–352. https://doi.org/10.1111/mam.12201 (3) Dobigny G, Morand S (2022) Zoonotic emergence at the animal-environment-human interface: the forgotten urban socio-ecosystems. Zenodo, 6444776, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.6444776 (4) Morand S, Lajaunie C (2021) Biodiversity and COVID-19: A report and a long road ahead to avoid another pandemic. One Earth, 4, 920–923. https://doi.org/10.1016/j.oneear.2021.06.007 | Zoonotic emergence at the animal-environment-human interface: the forgotten urban socio-ecosystems | Dobigny, G. & Morand, S. | <p style="text-align: justify;">Zoonotic emergence requires spillover from animals to humans, hence animal-human interactions. A lot has been emphasized on human intrusion into wild habitats (e.g., deforestation, hunting) and the development of ag... | | Disease Ecology/Evolution, Ecohealth, Ecology of hosts, infectious agents, or vectors, Evolution of hosts, infectious agents, or vectors, One Health, Zoonoses | Etienne Waleckx | 2022-04-11 11:39:11 | ||
06 Apr 2023

Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populationsChanges in aggressiveness in pathotypes of wheat leaf rustRecommended by Pierre Gladieux based on reviews by 2 anonymous reviewersUnderstanding the ecological and evolutionary factors underlying the spread of new fungal pathogen populations can inform the development of more effective management strategies. In plant pathology, pathogenicity is generally presented as having two components: ‘virulence’ (qualitative pathogenicity) and aggressiveness (quantitative pathogenicity). Changes in virulence in response to the deployment of new resistant varieties are a major driver of the spread of new populations (called pathotypes, or races) in modern agrosystems, and the genomic (i.e. proximal) and eco-evolutionary (i.e. ultimate) factors underlying these changes are well-documented [1,2,3]. By contrast, the role of changes in aggressiveness in the spread of pathotypes remains little known [4]. The study by Cécilia Fontyn and collaborators [5] set out to characterize changes in aggressiveness for isolates of two pathotypes of the wheat leaf rust (Puccinia triticina) that have been dominant in France during the 2005-2016 period. Isolates were genetically characterized using multilocus microsatellite typing and phenotypically characterized for three components of aggressiveness on wheat varieties: infection efficiency, latency period, and sporulation capacity. Using experiments that represent quite a remarkable amount of work and effort, Fontyn et al. showed that each dominant pathotype consisted of several genotypes, including common genotypes whose frequency changed over time. For each pathotype, the genotypes that were more common initially were replaced by a more aggressive genotype. Together, these results show that changes in the genetic composition of populations of fungal plant pathogens can be associated with, and may be caused by, changes in the quantitative components of pathogenicity. This study also illustrates how extensive, decade-long monitoring of fungal pathogen populations, such as the one conducted for wheat leaf rust in France, represents a very valuable resource for research. REFERENCES [1] Brown, J. K. (1994). Chance and selection in the evolution of barley mildew. Trends in Microbiology, 2(12), 470-475. https://doi.org/10.1016/0966-842x(94)90650-5 [2] Daverdin, G., Rouxel, T., Gout, L., Aubertot, J. N., Fudal, I., Meyer, M., Parlange, F., Carpezat, J., & Balesdent, M. H. (2012). Genome structure and reproductive behaviour influence the evolutionary potential of a fungal phytopathogen. PLoS Pathogens, 8(11), e1003020. https://doi.org/10.1371/journal.ppat.1003020 [3] Gladieux, P., Feurtey, A., Hood, M. E., Snirc, A., Clavel, J., Dutech, C., Roy, M., & Giraud, T. (2015). The population biology of fungal invasions.Molecular Ecology, 24(9), 1969-86. https://doi.org/10.1111/mec.13028 [4] Fontyn, C., Zippert, A. C., Delestre, G., Marcel, T. C., Suffert, F., & Goyeau, H. (2022). Is virulence phenotype evolution driven exclusively by Lr gene deployment in French Puccinia triticina populations?. Plant Pathology, 71(7), 1511-1524. https://doi.org/10.1111/ppa.13599 [5] Fontyn, C., Meyer, K. J., Boixel, A. L., Delestre, G., Piaget, E., Picard, C., Suffer, F., Marcel, T.C., & Goyeau, H. (2022). Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations. bioRxiv, 2022.08.29.505401, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.29.505401 | Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations | Cécilia FONTYN, Kevin JG MEYER, Anne-Lise BOIXEL, Ghislain DELESTRE, Emma PIAGET, Corentin PICARD, Frédéric SUFFERT, Thierry C MARCEL, Henriette GOYEAU | <p style="text-align: justify;">Plant pathogens are constantly evolving and adapting to their environment, including their host. Virulence alleles emerge, and then increase, and sometimes decrease in frequency within pathogen populations in respon... | | Coevolution, Epidemiology, Evolution of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Pathogenic/Symbiotic Fungi, Phytopathology, Plant diseases, Population dynamics of hosts, infectious agents, or... | Pierre Gladieux | Emerson Del Ponte , Jacqui Shykoff, Leïla Bagny Beilhe , Alexey Mikaberidze | 2022-09-29 20:01:57 | |
02 Jun 2023
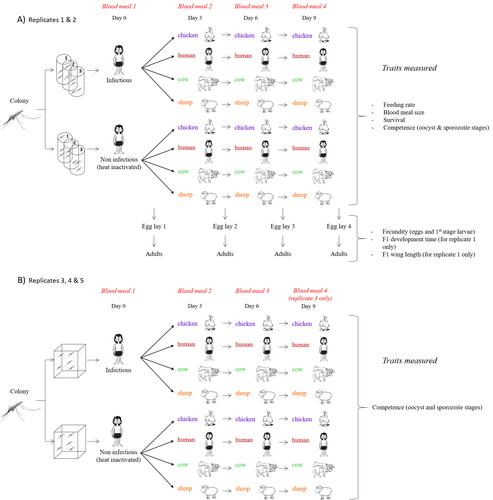
Multiple hosts, multiple impacts: the role of vertebrate host diversity in shaping mosquito life history and pathogen transmissionWhat you eat can eliminate you: bloodmeal sources and mosquito fitnessRecommended by Diego Santiago-Alarcon based on reviews by Francisco C. Ferreira and 1 anonymous reviewerDiptera-borne pathogens rank among the most serious health threats to vertebrate organisms around the world, particularly in tropical areas undergoing strong human impacts – e.g., urbanization and farming –, where social unrest and poor economies exacerbate the risk (Allen et al. 2017; Robles-Fernández et al. 2022). Although scientists have acquired a detailed knowledge on the life-history of malaria parasites (Pacheco and Escalante 2023), they still do not have enough information about their insect vectors to make informed management and preventive decisions (Santiago-Alarcon 2022). In this sense, I am pleased to recommend the study of Vantaux et al. (2023), where authors conducted an experimental and theoretical study to analyzed how the diversity of blood sources (i.e., human, cattle, sheep, and chicken) affected the fitness of the human malaria parasite – Plasmodium falciparum – and its mosquito vector – Anopheles gambiae s.l. The study was conducted in Burkina Faso, West Africa. Interestingly, authors did not find a significant effect of blood meal source on parasite development, and a seemingly low impact on the fitness of mosquitoes that were exposed to parasites. However, mosquitoes’ feeding rate, survival, fecundity, and offspring size were negatively affected by the type of blood meal ingested. In general, chicken blood represented the worst meal source for the different measures of mosquito fitness, and sheep blood seems to be the least harmful. This result was supported by the theoretical model, where vectorial capacity was always better when mosquitoes fed on sheep blood compared to cow and chicken blood. Thus, the knowledge generated by this study provides a pathway to reduce human infection risk by managing the diversity of farm animals. For instance, transmission to humans can decrease when chickens and cows represent most of the available blood sources in a village. These results along with other interesting details of this study, represent a clear example of the knowledge and understanding of insect vectors that we need to produce in the future, particularly to manage and prevent hazards and risks (sensu Hoseini et al. 2017). REFERENCES Allen T., et al., Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 8, 1124. (2017). https://doi.org/10.1038/s41467-017-00923-8 Hosseini P.R., et al., Does the impact of biodiversity differ between emerging and endemic pathogens? The need to separate the concepts of hazard and risk. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160129 (2017). https://doi.org/10.1098/rstb.2016.0129 Pacheco M.A., and Escalante, A.A., Origin and diversity of malaria parasites and other Haemosporida. Trend. Parasitol. (2023) https://doi.org/10.1016/j.pt.2023.04.004 Robles-Fernández A., et al., Wildlife susceptibility to infectious diseases at global scales. PNAS 119: e2122851119. (2022). https://doi.org/10.1073/pnas.2122851119 Santiago-Alarcon D. A meta-analytic approach to investigate mosquitoes’ (Diptera: Culicidae) blood feeding preferences from non-urban to urban environments. In: Ecology and Control of Vector-borne Diseases, vol. 7 (R.G. Gutiérrez-López, J.G. Logan, Martínez-de la Puente J., Eds). Pp. 161-177. Wageningen Academic Publishers. eISBN: 978-90-8686-931-2 | ISBN: 978-90-8686-379-2 (2022). Vantaux A. et al. Multiple hosts, multiple impacts: the role of vertebrate host diversity in shaping mosquito life history and pathogen transmission. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Infections (2023). https://doi.org/10.1101/2023.02.10.527988 | Multiple hosts, multiple impacts: the role of vertebrate host diversity in shaping mosquito life history and pathogen transmission | Amélie Vantaux, Nicolas Moiroux, Kounbobr Roch Dabiré, Anna Cohuet, Thierry Lefèvre | <p style="text-align: justify;">The transmission of malaria parasites from mosquito to human is largely determined by the dietary specialization of <em>Anopheles mosquitoes</em> to feed on humans. Few studies have explored the impact of blood meal... | | Ecology of hosts, infectious agents, or vectors, Parasites, Vectors | Diego Santiago-Alarcon | 2023-02-13 11:02:58 | ||
28 May 2024
HIV self-testing positivity rate and linkage to confirmatory testing and care: a telephone survey in Côte d'Ivoire, Mali and SenegalThe benefits of HIV self-testing in West Africa: quantified.Recommended by Jessie Abbate based on reviews by 3 anonymous reviewersDespite decades of advances and understanding of the indiscriminate nature of human immunodeficiency virus (HIV), it remains shrouded in stigma that makes it difficult to reach some key populations at risk of transmission. The advent of self-testing technology for HIV (HIVST) has opened much-needed potential for bringing privacy to prevention that is crucial for curtailing its continued spread (Johnson et al., 2014). The HIV Self-Testing in Africa (STAR) Initiative (https://www.psi.org/fr/project/star/), carried out in Eastern and Southern Africa between 2015 and 2020 (Simwinga et al., 2022), demonstrated the market and public health operational potential of HIVST of different distribution methods. From 2019 to 2022, the “AutoTest de dépistage du VIH : Libre d’Accéder à la connaissance de son Statut" (ATLAS, translating to “HIVST: Freedom to know your status”) program built on these findings to quantify the public health value of HIVST for reaching key populations in West Africa (specifically, Mali, Senegal and Côte d’Ivoire) (Ky-Zerbo et al., 2022). The innovative secondary distribution methods these studies employed, where the primary targeted populations were also encouraged to take and provide tests to their contacts, helped widen the reach of HIVST within key population networks beyond those relying on access to HIV testing facilities.
The tricky part of the self-testing model lies in assessing its reach and impact while maintaining the privacy of self-testers that is central to its success. Following voluntary phone survey methods that previously were able to show expanded reach of HIVST to first-time testers in key populations in West Africa and high rates of confirmatory testing and treatment seeking (Kra et al., 2022), Kra et al. (Kra et al., 2024) quantified how many of these self-tests led to a positive result – allowing wider assessment of follow-up behaviors and positivity rates among the hard-to-reach populations the program had targeted.
While the numbers were low, the results were informative. Among respondents who reported a positive (“reactive”) HIVST, just 44% proceeded to confirmatory testing. This is lower than in other populations where HIVST follow-up has been assessed (Thirumurthy et al., 2016). The main reasons given for not confirming a reactive self-test was misinterpretation of HIVST results and not understanding that confirmatory testing was needed. The result thus highlighted a need for improved communication on how to correctly interpret HIVST results, and the authors provided ranges for how this misinterpretation could have affected their positivity estimates. However, the majority of those who sought confirmatory testing did so within 3 months, and nearly all of those with confirmed infection started on treatment. HIV positivity rates in the three countries were all higher than other published HIV positivity estimates (Giguère et al., 2021; Maheu-Giroux et al., 2019), suggesting that HIVST methods were highly effective at reaching the targeted communities. Finally, while the authors demonstrated their methods as an effective way of assessing the utility of HIVST campaigns and identifying ways to improve them, the follow-up surveys are likely too costly to replace current passive surveillance methods for assessing community disease burden. That said, these precious data should be taken as validation of the public health value of HIV self-testing in key populations across communities in West Africa. With improvements in communicating instructions for use and follow-up, there is little doubt that the innovation of HIVST primary and secondary distribution could become a widely useful addition to the fight against HIV.
References Giguère, K., Eaton, J. W., Marsh, K., Johnson, L. F., Johnson, C. C., Ehui, E., Jahn, A., Wanyeki, I., Mbofana, F., Bakiono, F., Mahy, M., & Maheu-Giroux, M. (2021). Trends in knowledge of HIV status and efficiency of HIV testing services in sub-Saharan Africa, 2000–20: a modelling study using survey and HIV testing programme data. The Lancet HIV, 8(5), e284–e293. https://doi.org/10.1016/S2352-3018(20)30315-5 Johnson, C., Baggaley, R., Forsythe, S., Van Rooyen, H., Ford, N., Napierala Mavedzenge, S., Corbett, E., Natarajan, P., & Taegtmeyer, M. (2014). Realizing the potential for HIV self-testing. In AIDS and Behavior (Vol. 18, Issue SUPPL. 4). Springer New York LLC. https://doi.org/10.1007/s10461-014-0832-x Kra, A. K., Fosto, A. S., N’guessan, K. N., Geoffroy, O., Younoussa, S., Kabemba, O. K., Gueye, P. A., Ndeye, P. D., Rouveau, N., Boily, M. C., Silhol, R., d’Elbée, M., Maheu-Giroux, M., Vautier, A., & Larmarange, J. (2022). Can HIV self-testing reach first-time testers? A telephone survey among self-test end users in Côte d’Ivoire, Mali, and Senegal. BMC Infectious Diseases, 22. https://doi.org/10.1186/s12879-023-08626-w Kra, A. K., Fotso, A. S., Rouveau, N., Maheu-Giroux, M., Boily, M.-C., Silhol, R., d’Elbée, M., Vautier, A., Lamarange, J., & the Atlas team. (2024). HIV self-testing positivity rate and linkage to confirmatory testing and care: a telephone survey in Côte d’Ivoire, Mali, and Senegal. MedRxiv, Ver. 4 Peer-Reviewed and Recommended by Peer Community in Infections, 2023.06.10.23291206. https://doi.org/https://doi.org/10.1101/2023.06.10.23291206 Ky-Zerbo, O., Desclaux, A., Boye, S., Maheu-Giroux, M., Rouveau, N., Vautier, A., Camara, C. S., Kouadio, B. A., Sow, S., Doumenc-Aidara, C., Gueye, P. A., Geoffroy, O., Kamemba, O. K., Ehui, E., Ndour, C. T., Keita, A., & Larmarange, J. (2022). “I take it and give it to my partners who will give it to their partners”: Secondary distribution of HIV self-tests by key populations in Côte d’Ivoire, Mali, and Senegal. BMC Infectious Diseases, 22. https://doi.org/10.1186/s12879-023-08319-4 Maheu-Giroux, M., Marsh, K., Doyle, C. M., Godin, A., Lanièce Delaunay, C., Johnson, L. F., Jahn, A., Abo, K., Mbofana, F., Boily, M. C., Buckeridge, D. L., Hankins, C. A., & Eaton, J. W. (2019). National HIV testing and diagnosis coverage in sub-Saharan Africa: A new modeling tool for estimating the “first 90” from program and survey data. AIDS, 33, S255–S269. https://doi.org/10.1097/QAD.0000000000002386 Simwinga, M., Gwanu, L., Hensen, B., Sigande, L., Mainga, M., Phiri, T., Mwanza, E., Kabumbu, M., Mulubwa, C., Mwenge, L., Bwalya, C., Kumwenda, M., Mubanga, E., Mee, P., Johnson, C. C., Corbett, E. L., Hatzold, K., Neuman, M., Ayles, H., & Taegtmeyer, M. (2022). Lessons learned from implementation of four HIV self-testing (HIVST) distribution models in Zambia: applying the Consolidated Framework for Implementation Research to understand impact of contextual factors on implementation. BMC Infectious Diseases, 22(Suppl 1). https://doi.org/10.1186/s12879-024-09168-5 Thirumurthy, H., Masters, S. H., Mavedzenge, S. N., Maman, S., Omanga, E., & Agot, K. (2016). Promoting male partner HIV testing and safer sexual decision making through secondary distribution of self-tests by HIV-negative female sex workers and women receiving antenatal and post-partum care in Kenya: a cohort study. The Lancet HIV, 3(6), e266–e274. https://doi.org/10.1016/S2352-3018(16)00041-2
| HIV self-testing positivity rate and linkage to confirmatory testing and care: a telephone survey in Côte d'Ivoire, Mali and Senegal | Kra Djuhe Arsene Kouassi, Arlette Simo Fotso, Nicolas Rouveau, Mathieu Maheu-Giroux, Marie-Claude Boily, Romain Silhol, Marc d'Elbee, Anthony Vautier, Joseph Larmarange, ATLAS Team | <p>HIV self-testing (HIVST) empowers individuals to decide when and where to test and with whom to share their results. From 2019 to 2022, the ATLAS program distributed ~ 400 000 HIVST kits in Côte d’Ivoire, Mali, and Senegal. It prioritised key p... | | Epidemiology | Jessie Abbate | 2023-06-16 16:40:51 | ||
29 Jan 2024
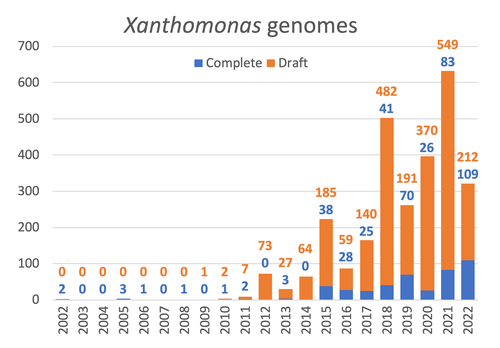
Celebrating the 20th Anniversary of the First Xanthomonas Genome Sequences – How Genomics Revolutionized Taxonomy, Provided Insight into the Emergence of Pathogenic Bacteria, Enabled New Fundamental Discoveries and Helped Developing Novel Control Measures – A Perspective from the French Network on XanthomonadsAdvancing Pathogen Genomics: A Comprehensive Review of the Xanthomonas(*) Genome's Impact on Bacterial Research and Control StrategiesRecommended by Damien François Meyer based on reviews by Boris Vinatzer and 3 anonymous reviewers based on reviews by Boris Vinatzer and 3 anonymous reviewers
The paper titled "Celebrating the 20th Anniversary of the First Xanthomonas Genome Sequences – How Genomics Revolutionized Taxonomy Provided Insight into the Emergence of Pathogenic Bacteria Enabled New Fundamental Discoveries and Helped Developing Novel Control Measures – A Perspective from the French Network on Xanthomonads" by Ralf Koebnik et al. (2023) is an insightful contribution to the field of genomics and its application in understanding pathogenic bacteria, particularly Xanthomonas. This comprehensive review offers a unique perspective from the French Network on Xanthomonads, underscoring the significant advancements in taxonomy, pathogen emergence, and development of control strategies due to genomic research. One of the paper's main strengths is its thorough exploration of how genomics has revolutionized our understanding of Xanthomonas and other pathogenic bacteria. It sheds light on the evolution and emergence of these pathogens, contributing significantly to the development of novel and effective control measures. The authors' detailed account of the historical progress and current state of genomics in this field highlights its pivotal role in guiding future research and practical applications in managing bacterial diseases. Moreover, the paper emphasizes the importance of collaborative efforts and the sharing of knowledge within scientific networks, as exemplified by the French Network on Xanthomonas. This approach not only enriches the study but also serves as a model for future collaborative research endeavors. In conclusion, the work of Koebnik et al. is a valuable resource for researchers, policymakers, and practitioners in the field of plant pathology and genomics. It not only provides a comprehensive overview of the advances in genomics related to Xanthomonas but also illustrates the broader impact of genomic studies in understanding and managing pathogenic bacteria. References Ralf Koebnik, Sophie Cesbron, Nicolas W. G. Chen, Marion Fischer-Le Saux, Mathilde Hutin, Marie-Agnès Jacques, Laurent D. Noël, Alvaro Perez-Quintero, Perrine Portier, Olivier Pruvost, Adrien Rieux, And Boris Szurek (2024) Celebrating the 20th anniversary of the first Xanthomonas genome gequences – How genomics revolutionized taxonomy, provided insight into the emergence of pathogenic bacteria, enabled new fundamental discoveries and helped developing novel control measures – A perspective from the French network on Xanthomonads. Zenodo ver. 3, peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.8223857 | Celebrating the 20th Anniversary of the First Xanthomonas Genome Sequences – How Genomics Revolutionized Taxonomy, Provided Insight into the Emergence of Pathogenic Bacteria, Enabled New Fundamental Discoveries and Helped Developing Novel Control ... | Ralf Koebnik, Sophie Cesbron, Nicolas W. G. Chen, Marion Fischer-Le Saux, Mathilde Hutin, Marie-Agnès Jacques, Laurent D. Noël, Alvaro Perez-Quintero, Perrine Portier, Olivier Pruvost, Adrien Rieux, And Boris Szurek | <p>In this Opinion paper, members of the French Network on Xanthomonads give their personal view on what they consider to be some of the groundbreaking discoveries in the field of molecular plant pathology over the past 20 years. By celebrating th... | | Epidemiology, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Molecular biology of infections, Molecular genetics o... | Damien François Meyer | 2023-08-09 10:37:15 | ||
23 Jan 2023

Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolenseWhole genome transcriptome reveals metabolic and immune susceptibility factors for Trypanosoma congolense infection in West-African livestockRecommended by Concepción Marañón based on reviews by 2 anonymous reviewersAfrican trypanosomiasis is caused by to the infection of a protozoan parasite of the Trypanosoma genus. It is transmitted by the tsetse fly, and is largely affecting cattle in the sub-humid areas of Africa, causing a high economic impact. However, not all the bovine strains are equally susceptible to the infection (1). In order to dissect the mechanisms underlying susceptibility to African trypanosoma infection, Peylhard et al (2) performed blood transcriptional profiles of trypanotolerant, trypanosensitive and mixed cattle breeds, before and after experimental infection with T. congolense. First of all, the authors have characterized the basal transcriptional profiles in the blood of the different breeds under study, which could be classified in a wide array of functional pathways. Of note, after infection some pathways were consistently enriched in all the group tested. Among them, the immune system-related ones were again on the top functions reported. The search for specific canonical pathways pointed to a prominent role of lipid and cholesterol-related pathways, as well as mitochondrial function and B and T lymphocyte activation. However, the analysis of infected animals demonstrated that trypanosusceptible animals showed a stronger transcriptomic reprogramming, highly enriched in specific metabolic and immunological pathways. It is worthy to highlight striking differences in genes involved in immune signal transduction, cytokines and markers of different leukocyte subpopulations. This work represents undoubtedly a significant momentum in the field, since the authors explore in deep a wide panel of cattle breeds representing the majority of West-African taurine and zebu in a systematic way. Since the animals were studied at different timepoints after infection, future longitudinal analyses of these datasets will be providing a precious insight on the kinetics of immune and metabolic reprogramming associated with susceptibility and tolerance to African trypanosoma infection, widening the application of this interesting study into new therapeutic interventions. References 1. Berthier D, Peylhard M, Dayo G-K, Flori L, Sylla S, Bolly S, Sakande H, Chantal I, Thevenon S (2015) A Comparison of Phenotypic Traits Related to Trypanotolerance in Five West African Cattle Breeds Highlights the Value of Shorthorn Taurine Breeds. PLOS ONE, 10, e0126498. https://doi.org/10.1371/journal.pone.0126498 2. Peylhard M, Berthier D, Dayo G-K, Chantal I, Sylla S, Nidelet S, Dubois E, Martin G, Sempéré G, Flori L, Thévenon S (2022) Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense. bioRxiv, 2022.06.10.495622, ver. 2 peer-reviewed and recommended by Peer Community Infections. https://doi.org/10.1101/2022.06.10.495622. | Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense | Moana Peylhard, David Berthier, Guiguigbaza-Kossigan Dayo, Isabelle Chantal, Souleymane Sylla, Sabine Nidelet, Emeric Dubois, Guillaume Martin, Guilhem Sempéré, Laurence Flori, Sophie Thévenon | <p>Animal African trypanosomosis, caused by blood protozoan parasites transmitted mainly by tsetse flies, represents a major constraint for millions of cattle in sub-Saharan Africa. Exposed cattle include trypanosusceptible indicine breeds, severe... | | Animal diseases, Genomics, functional genomics of hosts, infectious agents, or vectors, Resistance/Virulence/Tolerance | Concepción Marañón | Anonymous, Anonymous | 2022-06-14 17:06:57 | |
16 Jul 2024
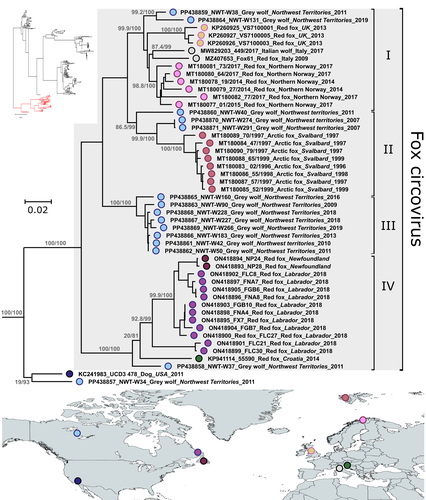
Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, CanadaWild canine viruses in the news. Better understanding multi-host transmission by adopting a disease ecology species community-based approachRecommended by Jean-Francois Guégan based on reviews by Arvind Varsani and 1 anonymous reviewerAccording to the international animal health authority, i.e., the World Organization on Animal Health (WOAH, former OIE), circoviruses are part of the Circoviridae family, which only includes 2 genera Circovirus and Cyclovirus, and infect swine, canine, ursid, viverrid, felid, pinniped, herpestid, mustelid, and several avian species (WOAH 2021). They are small (12–27 nm), non-enveloped, circular, single-stranded DNA viruses, viral replication is nuclear, and wild and domestic birds and mammals could serve as natural hosts. If most infections caused by circoviruses are subclinical in both wild and domestic species, they can be responsible for severe diseases in the commercial pig industry due to the Porcine circovirus-2 (PCV-2). These viruses can constitute a threat to wildlife, and cause their hosts to become immunocompromised, and animals often present with secondary coinfections. Canine circoviruses (CanineCV) harbour a worldwide distribution in dogs, and is the sole member of the viral genus to infect canines. They can be detected in wild carnivores, such as wolves, badgers, foxes and jackals, which indicates an ability for cross-species transmission between wildlife and domestic dogs. However, fox circovirus (FoCV), a distinct lineage of CanineCV, has been identified exclusively in wild canids (foxes and wolves) and not in dogs in Europe and North America, where it can cause in red foxes meningoencephalitis and other central nervous system signs. In their article, Canuti et al. (2024) investigate the presence, distribution and ecology of CanineCV in grey wolf specimens from the Northwest Territories, Canada. CanineCV occurrence appears to be relatively high with 45.3% positive specimens and parvoviral superinfections observed. The authors identify a high CanineCV genetic diversity among the investigated grey wolf specimens, and exacerbated by viral recombination. Phylogenetic analysis reveals the existence of 4 lineages, within each of them strains segregate by geography and not by host origin. This observed geographic segregation is interpreted as being due to the absence of exchange flows between grey wolf host subpopulations. Due to the paucity of knowledge on these circoviruses in wildlife and at the interface between wild and domestic animals, the authors discuss the plausible role of wolves as natural host reservoirs for disease transmission due to long-lasting virus-host coevolution. They are also conscious that additional maintenance hosts could exist in the wild, claiming for further studies to decipher fox circovirus disease ecology and transmission dynamics. This study underlines the importance of better understanding the transmission ecology and evolution of these Canine circoviruses, and I can only agree. Xiao et al. (2023), a research not referred to in the present work, evidenced CanineCV infection in cats in China, and obtained the first whole genome of cat-derived CanineCV. This emphasizes the importance of monitoring additional animal species and locations in the world to clarify disease ecology and transmission dynamics. A broader sampling of a wide range of animal species in different parts of the world using a species community-based approach is the key to understanding these CanineCV infections. References Marta CANUTI, Abigail V.L. KING, Giovanni FRANZO, H. Dean CLUFF, Lars E. LARSEN, Heather FENTON, Suzanne C. DUFOUR, Andrew S. LANG. 2024. Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, Canada. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2024.03.08.584028 World Organization on Animal Health. 2021. Circoviruses. https://www.woah.org/app/uploads/2021/05/circoviruses-infection-with.pdf [consulted on July 9th, 2024]. Xiangyu XIAO, Yan CHAO LI, Feng PEI XU, Xiangpi HAO, Shoujun LI, Pei ZHOU. 2023. Canine circovirus among dogs and cats in China: first identification in cats. Front. Microbiol. 14. https://doi.org/10.3389/fmicb.2023.1252272 | Diverse fox circovirus (*Circovirus canine*) variants circulate at high prevalence in grey wolves (*Canis lupus*) from the Northwest Territories, Canada | Marta Canuti, Abigail V.L. King, Giovanni Franzo, H. Dean Cluff, Lars E. Larsen, Heather Fenton, Suzanne C. Dufour, Andrew S. Lang | <p style="text-align: justify;">Canine circoviruses (CanineCV) have a worldwide distribution in dogs and are occasionally detected in wild carnivorans, indicating their ability for cross-species transmission. However, fox circovirus, a lineage of ... | | Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Epidemiology, Molecular genetics of hosts, infectious agents, or vectors, Population genetics of hosts, infectious agents, or vectors, Reservoirs, Taxonomy of hosts, infec... | Jean-Francois Guégan | Martine Peeters, Arvind Varsani | 2024-03-09 09:04:29 |




