
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender▼ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
03 Nov 2023

Longitudinal Survey of Astrovirus infection in different bat species in Zimbabwe: Evidence of high genetic Astrovirus diversityHigh diversity and evidence for inter-species transmission in astroviruses surveyed from bats in ZibabwaeRecommended by Tim James based on reviews by 2 anonymous reviewersMost infectious diseases of humans are zoonoses, and many of these come from particularly species diverse reservoir taxa, such as bats, birds, and rodents (1). Because of our changing landscape, there is increased exposure of humans to wildlife diseases reservoirs, yet we have little basic information about prevalence, hotspots, and transmission factors of most zoonotic pathogens. Viruses are particularly worrisome as a public health risk due to their fast mutation rates and well-known cross-species transmission abilities. There is a global push to better survey wildlife for viruses (2), but these studies are difficult, and the problem is vast. Astroviruses (AstVs) comprise a diverse family of ssRNA viruses known from mammals and birds. Astroviruses can cause gastroenteritis in humans and are more common in elderly and young children, but the relationship of human to non-human Astroviridae as well as transmission routes are unclear. AstVs have been detected at high prevalence in bats in multiple studies (3,4), but it is unclear what factors, such as co-infecting viruses and bat reproductive phenology, influence viral shedding and prevalence. References 1. Mollentze N, Streicker DG. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proceedings of the National Academy of Sciences. 2020 Apr 28;117(17):9423-30. https://doi.org/10.1073/pnas.1919176117 | Longitudinal Survey of Astrovirus infection in different bat species in Zimbabwe: Evidence of high genetic Astrovirus diversity | Vimbiso Chidoti, Helene De Nys, Malika Abdi, Getrudre Mashura, Valerie Pinarello, Ngoni Chiweshe, Gift Matope, Laure Guerrini, Davies Pfulenyi, Julien Cappelle, Ellen Mwandiringana, Dorothee Misse, Gori Elizabeth, Mathieu Bourgarel, Florian Liegeois | <p>Astroviruses (AstVs) have been discovered in over 80 animal species including diverse bat species and avian species. A study on Astrovirus circulation and diversity in different insectivorous and frugivorous chiropteran species roosting in tree... | | Animal diseases, Epidemiology, Molecular genetics of hosts, infectious agents, or vectors, Reservoirs, Viruses, Zoonoses | Tim James | 2023-04-18 14:58:43 | ||
14 Nov 2022
Ehrlichia ruminantium uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cellsAdhesion process of Ehrlichia ruminantium to its host cell: the role of the protein ERGACDS01230 elucidatedRecommended by Thomas Pollet based on reviews by Rodolfo García-Contreras and Alejandro Cabezas-CruzAs recently reported by the world organisation for animal health, 60% of infectious diseases are zoonotic with a significant part associated to ticks. Ticks can transmit various pathogens such as bacteria, viruses and parasites. Among pathogens known to be transmitted by ticks, Ehrlichia ruminantium is an obligate intracellular Gram-negative bacterium responsible for the fatal heartwater disease of domestic and wild ruminants (Allsopp, 2010). E. ruminantium is transmitted by ticks of the genus Amblyomma in the tropical and sub-Saharan areas, as well as in the Caribbean islands. It constitutes a major threat for the American livestock industries since a suitable tick vector is already present in the American mainland and potential introduction of infected A. variegatum through migratory birds or uncontrolled movement of animals from Caribbean could occur (i.e. Deem, 1998 ; Kasari et al 2010). The disease is also a major obstacle to the introduction of animals from heartwater-free to heartwater-infected areas into sub-Saharan Africa and thus restrains breeding programs aiming at upgrading local stocks (Allsopp, 2010). In this context, it is essential to develop control strategies against heartwater, as developing effective vaccines, for instance. Such an objective requires a better understanding of the early interaction of E. ruminantium and its host cells and of the mechanisms associated with bacterial adhesion to the host-cell. In this study, the authors. studied the role of E. ruminantium membrane protein ERGA_CDS_01230 in the adhesion process to host bovine aortic endothelial cells (BAEC). After successfully producing the recombinant version of the protein, Pinarello et al (2022) followed the in vitro culture of E. ruminantium in BAEC and observed that the expression of the protein peaked at the extracellular infectious elementary body stages. This result would suggest the likely involvement of the protein in the early interaction of E. ruminantium with its host cells. The authors then showed using flow cytometry, and scanning electron microscopy, that beads coated with the recombinant protein adhered to BAEC. In addition, they also observed that the adhesion protein of E. ruminantium interacted with proteins of the cell's lysate, membrane and organelle fractions. Additionally, enzymatic treatment, degrading dermatan and chondroitin sulfates on the surface of BAEC, was associated with a 50% reduction in the number of bacteria in the host cell after a development cycle, indicating that glycosaminoglycans might play a role in the adhesion of E. ruminantium to the host-cell. Finally, the authors observed that the adhesion protein of E. ruminantium induced a humoral response in vaccinated animals, making this protein a possible vaccine candidate. As rightly pointed out by both reviewers, the results of this study represent a significant advance (i) in the understanding of the role of the E. ruminantium membrane protein ERGA_CDS_01230 in the adhesion process to the host-cell and (ii) in the development of new control strategies against heartwater as this protein might potentially be used as an immunogen for the development of future vaccines. References Allsopp, B.A. (2010). Natural history of Ehrlichia ruminantium. Vet Parasitol 167, 123-135. https://doi.org/10.1016/j.vetpar.2009.09.014 Deem, S.L. (1998). A review of heartwater and the threat of introduction of Cowdria ruminantium and Amblyomma spp. ticks to the American mainland. J Zoo Wildl Med 29, 109-113. Kasari, T.R. et al (2010). Recognition of the threat of Ehrlichia ruminantium infection in domestic and wild ruminants in the continental United States. J Am Vet Med Assoc. 237:520-30. https://doi.org/10.2460/javma.237.5.520 Pinarello V, Bencurova E, Marcelino I, Gros O, Puech C, Bhide M, Vachiery N, Meyer DF (2022) Ehrlichia ruminantium uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cells. bioRxiv, 2021.06.15.447525, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2021.06.15.447525 | *Ehrlichia ruminantium* uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cells | Valérie Pinarello, Elena Bencurova, Isabel Marcelino, Olivier Gros, Carinne Puech, Mangesh Bhide, Nathalie Vachiery, Damien F. Meyer | <p><em>Ehrlichia ruminantium</em> is an obligate intracellular bacterium, transmitted by ticks of the genus <em>Amblyomma</em> and responsible for heartwater, a disease of domestic and wild ruminants. High genetic diversity of <em>E. ruminantium</... | | Interactions between hosts and infectious agents/vectors, Microbiology of infections | Thomas Pollet | Rodolfo García-Contreras, Alejandro Cabezas-Cruz | 2021-10-14 16:54:54 | |
07 Feb 2023

Three-way relationships between gut microbiota, helminth assemblages and bacterial infections in wild rodent populationsUnveiling the complex interactions between members of gut microbiomes: a significant advance provided by an exhaustive study of wild bank volesRecommended by Thomas Pollet based on reviews by Jason Anders and 1 anonymous reviewerThe gut of vertebrates is a host for hundreds or thousands of different species of microorganisms named the gut microbiome. This latter may differ greatly in natural environments between individuals, populations and species (1). The vertebrate gut microbiome plays key roles in host fitness through functions including nutrient acquisition, immunity and defense against infectious agents. While bank voles are small mammals potentially reservoirs of a large number of infectious agents, questions about the links between their gut microbiome and the presence of pathogens are scarcely addressed. In this study, Bouilloud et al. (2) used complementary analyses of community and microbial ecology to (i) assess the variability of gut bacteriome diversity and composition in wild populations of the bank vole Myodes glareolus collected in four different sites in Eastern France and (ii) evaluate the three-way interactions between the gut bacteriota, the gastro-intestinal helminths and pathogenic bacteria detected in the spleen. Authors identified important variations of the gut bacteriota composition and diversity among bank voles mainly explained by sampling localities. They found positive correlations between the specific richness of both the gut bacteria and the helminth community, as well as between the composition of these two communities, even when accounting for the influence of geographical distance. The helminths Aonchotheca murissylvatici, Heligmosomum mixtum and the bacteria Bartonella sp were the main taxa associated with the whole gut bacteria composition. Besides, changes in relative abundance of particular gut bacterial taxa were specifically associated with other helminths (Mastophorus muris, Catenotaenia henttoneni, Paranoplocephala omphalodes and Trichuris arvicolae) or pathogenic bacteria. Infections with Neoehrlichia mikurensis, Orientia sp, Rickettsia sp and P. omphalodes were especially associated with lower relative abundance of members of the family Erysipelotrichaceae (Firmicutes), while coinfections with higher number of bacterial infections were associated with lower relative abundance of members of the Bacteroidales family (Bacteroidetes). As pointed out by both reviewers, this study represents a significant advance in the field. I would like to commend the authors for this enormous work. The amount of data, analyses and results is considerable which has sometimes complicated the understanding of the story at the beginning of the evaluation process. Thanks to constructive scientific interactions with both reviewers through the two rounds of evaluation, the authors have efficiently addressed the reviewer's concerns and improved the manuscript, making this great story easier to read. The innovative results of this study emphasize the complex interlinkages between gut bacteriome and infections in wild animal populations and I strongly recommend this article for publication In Peer Community Infections. References (1) Vujkovic-Cvijin I, Sklar J, Jiang L, Natarajan L, Knight R, Belkaid Y (2020) Host variables confound gut microbiota studies of human disease. Nature, 587, 448–454. https://doi.org/10.1038/s41586-020-2881-9 (2) Bouilloud M, Galan M, Dubois A, Diagne C, Marianneau P, Roche B, Charbonnel N (2023) Three-way relationships between gut microbiota, helminth assemblages and bacterial infections in wild rodent populations. biorxiv, 2022.05.23.493084, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.05.23.493084 | Three-way relationships between gut microbiota, helminth assemblages and bacterial infections in wild rodent populations | Marie Bouilloud, Maxime Galan, Adelaide Dubois, Christophe Diagne, Philippe Marianneau, Benjamin Roche, Nathalie Charbonnel | <p>Background</p> <p>Despite its central role in host fitness, the gut microbiota may differ greatly between individuals. This variability is often mediated by environmental or host factors such as diet, genetics, and infections. Recently, a part... | | Disease Ecology/Evolution, Ecohealth, Interactions between hosts and infectious agents/vectors, Reservoirs, Zoonoses | Thomas Pollet | 2022-05-25 10:13:23 | ||
19 Jul 2023
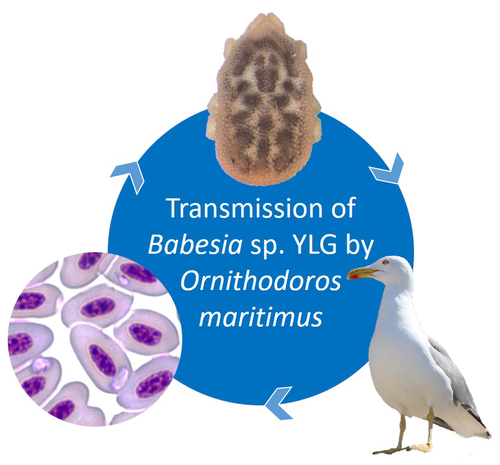
A soft tick vector of Babesia sp. YLG in Yellow-legged gull (Larus michahellis) nestsA four-year study reveals the potential role of the soft tick Ornithodoros maritimus in the transmission and circulation of Babesia sp. YLG in Yellow-legged gull colonies.Recommended by Thomas Pollet based on reviews by Hélène Jourdan and Tahar KernifWorldwide, ticks and tick-borne diseases are a persistent example of problems at the One Health interface between humans, wildlife, and environment (1, 2). The management and prevention of ticks and tick-borne diseases require a better understanding of host, tick and pathogen interactions and thus get a better view of the tick-borne pathosystems. In this study (3), the tick-borne pathosystem included three component species: first a seabird host, the Yellow-legged gull (YLG - Larus michahellis, Laridae), second a soft nidicolous tick (Ornithodoros maritimus, Argasidae, syn. Alectorobius maritimus) known to infest this host and third a blood parasite (Babesia sp. YLG, Piroplasmidae). In this pathosystem, authors investigated the role of the soft tick, Ornithodoros maritimus, as a potential vector of Babesia sp. YLG. They analyzed the transmission of Babesia sp. YLG by collecting different tick life stages from YLG nests during 4 consecutive years on the islet of Carteau (Gulf of Fos, Camargue, France). Ticks were dissected and organs were analyzed separately to detect the presence of Babesia sp DNA and to evaluate different transmission pathways. While the authors detected Babesia sp. YLG DNA in the salivary glands of nymphs, females and males, this result reveals a strong suspicion of transmission of the parasite by the soft tick. Babesia sp. YLG DNA was also found in tick ovaries, which could indicate possible transovarial transmission. Finally, the authors detected Babesia sp. YLG DNA in several male testes and in endospermatophores, and notably in a parasite-free female (uninfected ovaries and salivary glands). These last results raise the interesting possibility of sexual transmission from infected males to uninfected females. As pointed out by both reviewers, this is a nice study, well written and easy to read. All the results are new and allow to better understand the role of the soft tick, Ornithodoros maritimus, as a potential vector of Babesia sp. YLG. They finally question about the degree to which the parasite can be maintained locally by ticks and the epidemiological consequences of infection for both O. maritimus and its avian host. For all these reasons, I chose to recommend this article for Peer Community In Infections. References
| A soft tick vector of *Babesia* sp. YLG in Yellow-legged gull (*Larus michahellis*) nests | Claire Bonsergent, Marion Vittecoq, Carole Leray, Maggy Jouglin, Marie Buysse, Karen D. McCoy, Laurence Malandrin | <p style="text-align: justify;"><em>Babesia </em>sp. YLG has recently been described in Yellow-legged gull (<em>Larus michahellis</em>) chicks and belongs to the Peircei clade in the new classification of Piroplasms. Here, we studied <em>Babesia <... | | Ecology of hosts, infectious agents, or vectors, Eukaryotic pathogens/symbionts, Interactions between hosts and infectious agents/vectors, Parasites, Vectors | Thomas Pollet | 2023-03-29 14:33:40 | ||
24 Jan 2024
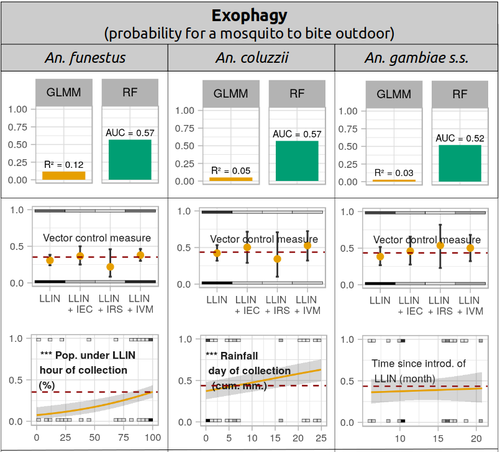
Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictabilityLarge and complete datasets, and modelling reveal the major determinants of physiological and behavioral insecticide resistance of malaria vectorsRecommended by Thierry DE MEEÛS based on reviews by Haoues Alout and 1 anonymous reviewer based on reviews by Haoues Alout and 1 anonymous reviewer
Parasites represent the most diverse and adaptable ecological group of the biosphere (Timm & Clauson, 1988; De Meeûs et al., 1998; Poulin & Morand, 2000; De Meeûs & Renaud, 2002). The human species is known to considerably alter biodiversity, though it hosts, and thus sustains the maintenance of a spectacular diversity of parasites (179 species for eukaryotic species only) (De Meeûs et al., 2009). Among these, the five species of malaria agents (genus Plasmodium) remain a major public health issue around the world. Plasmodium falciparum is the most prevalent and lethal of these (Liu et al., 2010). With a pick of up to 2 million deaths due to malaria in 2004, deaths decreased to around 1 million in 2010 (Murray et al., 2012), to reach 619,000 in 2021, most of which in sub-Saharan Africa, and 79% of which were among children aged under 5 years (World Health Organization, 2022). As stressed by Taconet et al. (2023), reduction in malaria deaths is attributable to control measures, in particular against its vectors (mosquitoes of the genus Anopheles). Nevertheless, the success of vector control is hampered by several factors (biological, environmental and socio-economic), and in particular by the great propensity of targeted mosquitoes to evolve physiological or behavioral avoidance of anti-vectorial measures. In their paper Taconet et al. (2023) aims at understanding what are the main factors that determine the evolution of insecticide resistance in several malaria vectors, in relation to the biological determinisms of behavioral resistance and how fast such evolutions take place. To tackle these objectives, authors collected an impressive amount of data in two rural areas of West Africa. With appropriate modeling, Taconet et al. discovered, among many other results, a predominant role of public health measures, as compared to agricultural practices, in the evolution of physiological resistance. They also found that mosquito foraging activities are mostly explained by host availability and climate, with a poor, if any, association with genetic markers of physiological resistance to insecticides. These findings represent an important contribution to the field and should help at designing more efficient control strategies against malaria.
References De Meeûs T, Michalakis Y, Renaud F (1998) Santa Rosalia revisited: or why are there so many kinds of parasites in “the garden of earthly delights”? Parasitology Today, 14, 10–13. https://doi.org/10.1016/S0169-4758(97)01163-0 De Meeûs T, Prugnolle F, Agnew P (2009) Asexual reproduction in infectious diseases. In: Lost Sex: The Evolutionary Biology of Parthenogenesis (eds Schön I, Martens K, van Dijk P), pp. 517-533. Springer, NY. https://doi.org/10.1007/978-90-481-2770-2_24 De Meeûs T, Renaud F (2002) Parasites within the new phylogeny of eukaryotes. Trends in Parasitology, 18, 247–251. https://doi.org/10.1016/S1471-4922(02)02269-9 Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF, Ndjango JB, Sanz CM, Morgan DB, Locatelli S, Gonder MK, Kranzusch PJ, Walsh PD, Delaporte E, Mpoudi-Ngole E, Georgiev AV, Muller MN, Shaw GM, Peeters M, Sharp PM, Rayner JC, Hahn BH (2010) Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature, 467, 420–425. https://doi.org/10.1038/nature09442 Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. The Lancet, 379, 413–431. https://doi.org/10.1016/S0140-6736(12)60034-8 Poulin R, Morand S (2000) The diversity of parasites. Quarterly Review of Biology, 75, 277–293. https://doi.org/10.1086/393500 Taconet P, Soma DD, Zogo B, Mouline K, Simard F, Koffi AA, Dabire RK, Pennetier C, Moiroux N (2023) Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictability. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.20.504631 Timm RM, Clauson BL (1988) Coevolution: Mammalia. In: 1988 McGraw-Hill yearbook of science & technology, pp. 212–214. McGraw-Hill Book Company, New York. World Health Organization (2022) World malaria report 2022. Geneva: World Health Organization; 2022. Licence: CC BY-NC-SA 3.0 IGO. https://iris.who.int/bitstream/handle/10665/365169/9789240064898-eng.pdf?sequence=1.
| Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictability | Paul Taconet, Dieudonné Diloma Soma, Barnabas Zogo, Karine Mouline, Frédéric Simard, Alphonsine Amanan Koffi, Roch Kounbobr Dabiré, Cédric Pennetier, Nicolas Moiroux | <p>Insecticide resistance and behavioural adaptation of malaria mosquitoes affect the efficacy of long-lasting insecticide nets - currently the main tool for malaria vector control. To develop and deploy complementary, efficient and cost-effective... | | Behaviour of hosts, infectious agents, or vectors, Ecology of hosts, infectious agents, or vectors, Pesticide resistance, Population genetics of hosts, infectious agents, or vectors, Vectors | Thierry DE MEEÛS | Haoues Alout, Anonymous | 2023-07-03 11:29:10 | |
14 Dec 2022

Transcriptome responses of the aphid vector Myzus persicae are shaped by identities of the host plant and the virusHow do multiple host plants and virus species challenge aphid molecular machinery?Recommended by Sebastien Massart based on reviews by Juan José Lopez Moya and Michelle HeckThe impact of virus infection of a plant on an aphid’s behaviour has been observed in many studies [1]. Indeed, virus infection can alter plant biochemistry through the emission of volatile organic compounds and plant tissue content modification. These alterations can further impact the interactions between plants and aphids. However, although it is a well-known phenomenon, very few studies have explored the consequences of plant virus infection on the gene expression of aphids to understand better the aphid’s manipulation by the plant virus. In this context, the recommended study [2] reports a comprehensive transcriptomic analysis of the genes expressed by one aphid species, Myzus persicae, a vector of several plant viruses, when feeding on plants. Michelle Heck underlined how significant this study is for comprehending the molecular bases of aphid-vector manipulation by plant viruses (see below). Interestingly, the study design has integrated several factors that might influence the gene expression of M. persicae when feeding on the plant. Indeed, the authors investigated the effect of two plant species (Arabidopsis thaliana and Camelia sativa) and two virus species [turnip yellows virus (TuYV) and cauliflower mosaic virus (CaMV)]. Noteworthy, the transmission mode of TuYV is circulative and persistent, while CaMV is transmitted by a semi-persistent non-circulative mode. As Juan José Lopez Moya mentioned, multiple comparisons allowed the identification of the different responses of aphids in front of different host plants infected or not by different viruses (see below). This publication is complementary to a previous publication from the same team focusing on plant transcriptome analysis [3]. Thanks to their experimental design, the authors identified genes commonly deregulated by both viruses and/or both plant species and deregulated genes by a single virus or a single plant. Figure 4 nicely summarizes the number of deregulated genes. A thorough discussion on the putative role of deregulated genes in different conditions gave a comprehensive follow-up of the results and their impact on the current knowledge of plant-virus-vector interactions. This study has now opened the gate to promising research focusing on the functional validation of the identified genes while also narrowing the study from the body to the tissue level. References: 1. Carr JP, Tungadi T, Donnelly R, Bravo-Cazar A, Rhee S-J, Watt LG, Mutuku JM, Wamonje FO, Murphy AM, Arinaitwe W, Pate AE, Cunniffe NJ, Gilligan CA (2020) Modelling and manipulation of aphid-mediated spread of non-persistently transmitted viruses. Virus Research, 277, 197845. https://doi.org/10.1016/j.virusres.2019.197845 2. Chesnais Q, Golyaev V, Velt A, Rustenholz C, Verdier M, Brault V, Pooggin MM, Drucker M (2022) Transcriptome responses of the aphid vector Myzus persicae are shaped by identities of the host plant and the virus. bioRxiv , 2022.07.18.500449, ver. 5 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.07.18.500449 3. Chesnais Q, Golyaev V, Velt A, Rustenholz C, Brault V, Pooggin MM, Drucker M (2022) Comparative Plant Transcriptome Profiling of Arabidopsis thaliana Col-0 and Camelina sativa var. Celine Infested with Myzus persicae Aphids Acquiring Circulative and Noncirculative Viruses Reveals Virus- and Plant-Specific Alterations Relevant to Aphid Feeding Behavior and Transmission. Microbiology Spectrum, 10, e00136-22. https://doi.org/10.1128/spectrum.00136-22 | Transcriptome responses of the aphid vector *Myzus persicae* are shaped by identities of the host plant and the virus | Quentin Chesnais, Victor Golyaev, Amandine Velt, Camille Rustenholz, Maxime Verdier, Véronique Brault, Mikhail M. Pooggin, Martin Drucker | <p style="text-align: justify;"><strong>Background:</strong> Numerous studies have documented modifications in vector orientation behavior, settling and feeding behavior, and/or fecundity and survival due to virus infection in host plants. These a... | | Behaviour of hosts, infectious agents, or vectors, Cell biology of hosts, infectious agents, or vectors, Molecular biology of infections, Physiology of hosts, infectious agents, or vectors, Phytopathology, Plant diseases, Vectors, Viruses | Sebastien Massart | 2022-07-19 15:24:14 | ||
27 Feb 2023
African army ants at the forefront of virome surveillance in a remote tropical forestA groundbreaking study using ants revealed a spectacular diversity of viruses in hardly accessible ecosystems like tropical forestsRecommended by Sebastien Massart based on reviews by Mart Krupovic and 1 anonymous reviewerDeciphering the virome (the set or assemblage of viruses) of the Earth, from individual organisms to entire ecosystems, has become a key priority. The first step to better understanding the impact of viruses on the ecology and functions of ecosystems is to describe their diversity. Such knowledge opens the gates to a better assessment of global nutrient cycling or of the threat that viruses represent to individual health. This explains the increasing number of pioneering studies that are currently sequencing the complete or partial genome of thousands of new viruses [1]. In their exciting study, Fritz and collaborators [2], authors sampled 209 army ants (Genus Dorylus) to investigate the virus diversity in dense forests that researchers cannot easily access. Indeed, these ants live in colonies (21 were sampled) that can move 1 km per day, covering a significant area and attacking many invertebrate and vertebrate preys. Each sample was sequenced by a protocol called VANA sequencing and allowing the enrichment of the sample in viral sequences [3], so improving the detection of viruses present at low abundance in the ant (and more specifically in its gut for viruses infecting preys). Around 45,000 contigs presented homologies with bacterial, plant, invertebrate, and vertebrate infecting viruses. Half could be assigned to 56 families and 157 genera of the International Committee on Taxonomy of Viruses. Beyond this amazing harvest of new and known virus sequences using an original methodology, the results significantly improve the current frontiers of known viral taxonomy and diversity and raise exciting research tracks to expand them. As a preprint, several blogs or news of leading scientists and journals have already highlighted this study. For example, in the news section of Science magazine, Jon Cohen underlined the originality of the approach for virus hunting on Earth with the title “Armed with air samplers, rope tricks, and—yes—ants, virus hunters spot threats in new ways”[4]. Another example is the mention of the publication by Elisabeth Bik in her Microbiome Digest: she wrote, “An amazing read is a fresh preprint from Fritz and collaborator describing an exciting method of sampling in difficult-to-reach environments“ [5]. The paper from Fritz et al [2] thus represents a significant advance in virus ecology, as already recognized by early readers, and this is why I strongly recommend its publication in PCI Infections. REFERENCES 1. Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, Lohr D, Novakovsky G, Buchfink B, Al-Shayeb B, Banfield JF, de la Peña M, Korobeynikov A, Chikhi R, Babaian A (2022) Petabase-scale sequence alignment catalyses viral discovery. Nature, 602, 142–147. https://doi.org/10.1038/s41586-021-04332-2 2. Fritz M, Reggiardo B, Filloux D, Claude L, Fernandez E, Mahé F, Kraberger S, Custer JM, Becquart P, Mebaley TN, Kombila LB, Lenguiya LH, Boundenga L, Mombo IM, Maganga GD, Niama FR, Koumba J-S, Ogliastro M, Yvon M, Martin DP, Blanc S, Varsani A, Leroy E, Roumagnac P (2023) African army ants at the forefront of virome surveillance in a remote tropical forest. bioRxiv, 2022.12.13.520061, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.12.13.520061 3. François S, Filloux D, Fernandez E, Ogliastro M, Roumagnac P (2018) Viral Metagenomics Approaches for High-Resolution Screening of Multiplexed Arthropod and Plant Viral Communities. In: Viral Metagenomics: Methods and Protocols Methods in Molecular Biology. (eds Pantaleo V, Chiumenti M), pp. 77–95. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7683-6_7 4. Cohen J (2023) Virus hunters test new surveillance tools. Science, 379, 16–17. https://doi.org/10.1126/science.adg5292 5. Ponsero A (2023) February 18th, 2023. Microbiome Digest - Bik’s Picks. https://microbiomedigest.com/2023/02/18/february-18th-2023/ | African army ants at the forefront of virome surveillance in a remote tropical forest | Matthieu Fritz, Berenice Reggiardo, Denis Filloux, Lisa Claude, Emmanuel Fernandez, Frederic Mahe, Simona Kraberger, Joy M. Custer, Pierre Becquart, Telstar Ndong Mebaley, Linda Bohou Kombila, Leadisaelle H. Lenguiya, Larson Boundenga, Illich M. M... | <p style="text-align: justify;">In this study, we used a predator-enabled metagenomics strategy to sample the virome of a remote and difficult-to-access densely forested African tropical region. Specifically, we focused our study on the use of arm... | | Ecohealth, Ecology of hosts, infectious agents, or vectors, One Health, Reservoirs, Viruses | Sebastien Massart | 2022-12-14 11:57:40 | ||
21 Sep 2023
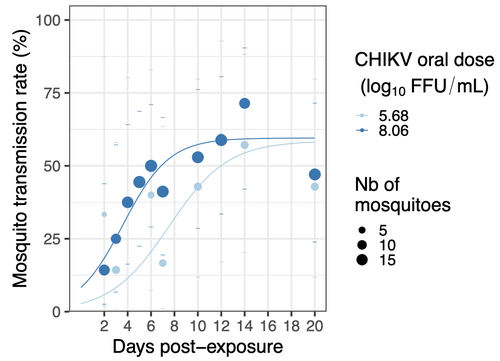
Chikungunya intra-vector dynamics in Aedes albopictus from Lyon (France) upon exposure to a human viremia-like dose range reveals vector barrier permissiveness and supports local epidemic potentialFill in one gap in our understanding of CHIKV intra-vector dynamicsRecommended by Sara Moutailler based on reviews by 2 anonymous reviewers
Mosquitoes are first vector of pathogen worldwide and transmit several arbovirus, most of them leading to major outbreaks (1). Chikungunya virus (CHIKV) is a perfect example of the “explosive type” of arbovirus, as observed in La Réunion Island in 2005-2006 (2-6) and also in the outbreak of 2007 in Italy (7), both vectorized by Ae. albopictus. Being able to better understand CHIKV intra-vector dynamics is still of major interest since not all chikungunya strain are explosive ones (8). In this study (9), the authors have evaluated the vector competence of a local strain of Aedes albopictus (collected in Lyon, France) for CHIKV. They evaluated infection, dissemination and transmission dynamics of CHIKV using different dose of virus in individual mosquitoes from day 2 to day 20 post exposure, by titration and quantification of CHIKV RNA load in the saliva. As highlighted by both reviewers, the most innovative idea in this study was the use of three different oral doses trying to span human viraemia detected in two published studies (10-11), doses that were estimated through their model of human CHIKV viremia in the blood. They have found that CHIKV dissemination from the Ae. albopictus midgut depends on the interaction between time post-exposure and virus dose (already highlighted by other international publications). Then their results were implemented in the agent-based model nosoi to estimate the epidemic potential of CHIKV in a French population of Ae. albopictus, using realistic vectorial capacity parameters. To conclude, the authors have discussed the importance of other parameters that could influence vector competence as mosquito microbiota and temperature, parameters that need also to be estimated in local mosquito population to improve the risk assessment through modelling. As pointed out by both reviewers, this is a nice study, well written and easy to read. These results allow filling in another gap of our understanding of CHIKV intra-vector dynamics and highlight the epidemic potential of CHIKV upon transmission by Aedes albopictus in mainland France. For all these reasons, I chose to recommend this article for Peer Community In Infections. References 1. Marine Viglietta, Rachel Bellone, Adrien Albert Blisnick, Anna-Bella Failloux. (2021). Vector Specificity of Arbovirus Transmission. Front Microbiol Dec 9;12:773211. https://doi.org/10.3389/fmicb.2021.773211 2. Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney M-C, Lavenir R, Pardigon N, Reynes J-M, Pettinelli F, Biscornet L, Diancourt L, Michel S, Duquerroy S, Guigon G, Frenkiel M-P, Bréhin A-C, Cubito N, Desprès P, Kunst F, Rey FA, Zeller H, Brisse S. (2006). Genome Microevolution of Chikungunya viruses Causing the Indian Ocean Outbreak. 2006. PLoS Medicine, 3, e263. https://doi.org/10.1371/journal.pmed.0030263 3. Bonilauri P, Bellini R, Calzolari M, Angelini R, Venturi L, Fallacara F, Cordioli P, 687 Angelini P, Venturelli C, Merialdi G, Dottori M. (2008). Chikungunya Virus in Aedes albopictus, Italy. Emerging Infectious 689 Diseases, 14, 852–854. https://doi.org/10.3201/eid1405.071144 4. Pagès F, Peyrefitte CN, Mve MT, Jarjaval F, Brisse S, Iteman I, Gravier P, Tolou H, Nkoghe D, Grandadam M. (2009). Aedes albopictus Mosquito: The Main Vector of the 2007 Chikungunya Outbreak in Gabon. PLoS ONE, 4, e4691. https://doi.org/10.1371/journal.pone.0004691 5. Paupy C, Kassa FK, Caron M, Nkoghé D, Leroy EM (2012) A Chikungunya Outbreak Associated with the Vector Aedes albopictus in Remote Villages of Gabon. Vector-Borne and Zoonotic Diseases, 12, 167–169. https://doi.org/10.1089/vbz.2011.0736 6. Mombouli J-V, Bitsindou P, Elion DOA, Grolla A, Feldmann H, Niama FR, Parra H-J, Munster VJ. (2013). Chikungunya Virus Infection, Brazzaville, Republic of Congo, 2011. Emerging Infectious Diseases, 19, 1542–1543. https://doi.org/10.3201/eid1909.130451 7. Venturi G, Luca MD, Fortuna C, Remoli ME, Riccardo F, Severini F, Toma L, Manso MD, Benedetti E, Caporali MG, Amendola A, Fiorentini C, Liberato CD, Giammattei R, Romi R, Pezzotti P, Rezza G, Rizzo C. (2017). Detection of a chikungunya outbreak in Central Italy, August to September 2017. Eurosurveillance, 22, 17–00646. https://doi.org/10.2807/1560-7917.es.2017.22.39.17-00646 8. de Lima Cavalcanti, T.Y.V.; Pereira, M.R.; de Paula, S.O.; Franca, R.F.d.O. (2022). A Review on Chikungunya Virus Epidemiology, Pathogenesis and Current Vaccine Development. Viruses 2022, 14, 969. https://doi.org/10.3390/v14050969 9. Barbara Viginier, Lucie Cappuccio, Celine Garnier, Edwige Martin, Carine Maisse, Claire Valiente Moro, Guillaume Minard, Albin Fontaine, Sebastian Lequime, Maxime Ratinier, Frederick Arnaud, Vincent Raquin. (2023). Chikungunya intra-vector dynamics in Aedes albopictus from Lyon (France) upon exposure to a human viremia-like dose range reveals vector barrier permissiveness and supports local epidemic potential. medRxiv, ver.3, peer-reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2022.11.06.22281997 10. Appassakij H, Khuntikij P, Kemapunmanus M, Wutthanarungsan R, Silpapojakul K (2013) Viremic profiles in CHIKV-infected cases. Transfusion, 53, 2567–2574. https://doi.org/10.1111/j.1537-2995.2012.03960.x 11. Riswari SF, Ma’roef CN, Djauhari H, Kosasih H, Perkasa A, Yudhaputri FA, Artika IM, Williams M, Ven A van der, Myint KS, Alisjahbana B, Ledermann JP, Powers AM, Jaya UA (2015) Study of viremic profile in febrile specimens of chikungunya in Bandung, Indonesia. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology, 74, 61–5. https://doi.org/10.1016/j.jcv.2015.11.017 | Chikungunya intra-vector dynamics in *Aedes albopictus* from Lyon (France) upon exposure to a human viremia-like dose range reveals vector barrier permissiveness and supports local epidemic potential | Barbara Viginier, Lucie Cappuccio, Celine Garnier, Edwige Martin, Carine Maisse, Claire Valiente Moro, Guillaume Minard, Albin Fontaine, Sebastian Lequime, Maxime Ratinier, Frederick Arnaud, Vincent Raquin | <p>Arbovirus emergence and epidemic potential, as approximated by the vectorial capacity formula, depends on host and vector parameters, including the vector intrinsic ability to replicate then transmit the pathogen known as vector competence. Vec... | | Epidemiology, Vectors, Viruses | Sara Moutailler | 2023-06-17 15:59:17 | ||
08 Aug 2023
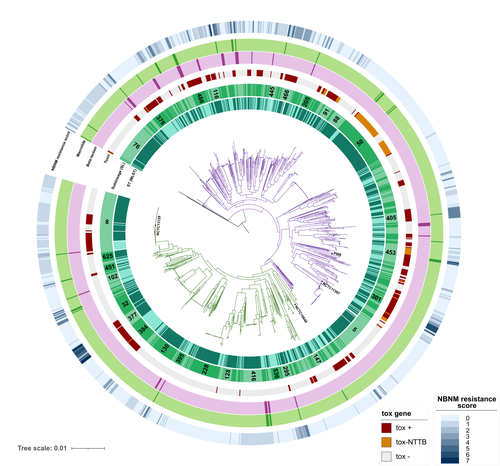
A global Corynebacterium diphtheriae genomic framework sheds light on current diphtheria reemergenceDIPHTOSCAN : A new tool for the genomic surveillance of diphtheriaRecommended by Rodolfo García-Contreras based on reviews by Ankur Mutreja and 2 anonymous reviewersOne of the greatest achievements of health sciences is the eradication of infectious diseases such as smallpox that in the past imposed a severe burden on humankind, through global vaccination campaigns. Moreover, progress towards the eradication of others such as poliomyelitis, dracunculiasis, and yaws is being made. In contrast, other infections that were previously contained are reemerging, due to several factors, including lack of access to vaccines due to geopolitical reasons, the rise of anti-vaccine movements, and the constant mobility of infected persons from the endemic sites. One of such disease is diphtheria, caused by Corynebacterium diphtheriae and a few other related species such as C. ulcerans and C. pseudotuberculosis. Importantly, in France, diphtheria cases reported in 2022 increased 7-fold from the average of previously recorded cases per year in the previous 4 years and the situation in other European countries is similar. Hence, as reported here, Hennart et al. (2023) developed DIPHTOSCAN, a free access bioinformatics tool with user-friendly interphase, aimed to easily identify, extract and interpret important genomic features such as the sublineage of the strain, the presence of the tox gene (as a string predictor for toxigenic disease) as well as genes coding other virulence factors such as fimbriae, and the presence of know resistant mechanisms towards antibiotics like penicillin and erythromycin currently used in the clinic to treat this infection. The authors validated the performance of their tool with a large collection of genomes, including those obtained from the isolates of the 2022 outbreak in France, more than 1,200 other genomes isolated from France, Algeria, and Yemen, and more than 500 genomes from several countries from Europe, America, Africa, Asia, and Oceania that are available through the NCBI site. DIPHTOSCAN will allow the rapid identification and surveillance of potentially dangerous strains such as those being tox-positive isolates and resistant to multiple drugs and/or first-line treatments and a better understanding of the epidemiology and evolution of this important reemerging disease. | A global *Corynebacterium diphtheriae* genomic framework sheds light on current diphtheria reemergence | Melanie Hennart, Chiara Crestani, Sebastien Bridel, Nathalie Armatys, Sylvie Brémont, Annick Carmi-Leroy, Annie Landier, Virginie Passet, Laure Fonteneau, Sophie Vaux, Julie Toubiana, Edgar Badell, Sylvain Brisse | <p style="text-align: justify;"><strong>Background</strong></p> <p style="text-align: justify;">Diphtheria, caused by <em>Corynebacterium diphtheriae</em>, reemerges in Europe since 2022. Genomic sequencing can inform on transmission routes and g... | | Drug resistance, tolerance and persistence, Epidemiology, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Microbiology of infections, Population genetics of hosts, infectiou... | Rodolfo García-Contreras | Ankur Mutreja | 2023-03-09 16:02:27 | |
06 Apr 2023

Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populationsChanges in aggressiveness in pathotypes of wheat leaf rustRecommended by Pierre Gladieux based on reviews by 2 anonymous reviewersUnderstanding the ecological and evolutionary factors underlying the spread of new fungal pathogen populations can inform the development of more effective management strategies. In plant pathology, pathogenicity is generally presented as having two components: ‘virulence’ (qualitative pathogenicity) and aggressiveness (quantitative pathogenicity). Changes in virulence in response to the deployment of new resistant varieties are a major driver of the spread of new populations (called pathotypes, or races) in modern agrosystems, and the genomic (i.e. proximal) and eco-evolutionary (i.e. ultimate) factors underlying these changes are well-documented [1,2,3]. By contrast, the role of changes in aggressiveness in the spread of pathotypes remains little known [4]. The study by Cécilia Fontyn and collaborators [5] set out to characterize changes in aggressiveness for isolates of two pathotypes of the wheat leaf rust (Puccinia triticina) that have been dominant in France during the 2005-2016 period. Isolates were genetically characterized using multilocus microsatellite typing and phenotypically characterized for three components of aggressiveness on wheat varieties: infection efficiency, latency period, and sporulation capacity. Using experiments that represent quite a remarkable amount of work and effort, Fontyn et al. showed that each dominant pathotype consisted of several genotypes, including common genotypes whose frequency changed over time. For each pathotype, the genotypes that were more common initially were replaced by a more aggressive genotype. Together, these results show that changes in the genetic composition of populations of fungal plant pathogens can be associated with, and may be caused by, changes in the quantitative components of pathogenicity. This study also illustrates how extensive, decade-long monitoring of fungal pathogen populations, such as the one conducted for wheat leaf rust in France, represents a very valuable resource for research. REFERENCES [1] Brown, J. K. (1994). Chance and selection in the evolution of barley mildew. Trends in Microbiology, 2(12), 470-475. https://doi.org/10.1016/0966-842x(94)90650-5 [2] Daverdin, G., Rouxel, T., Gout, L., Aubertot, J. N., Fudal, I., Meyer, M., Parlange, F., Carpezat, J., & Balesdent, M. H. (2012). Genome structure and reproductive behaviour influence the evolutionary potential of a fungal phytopathogen. PLoS Pathogens, 8(11), e1003020. https://doi.org/10.1371/journal.ppat.1003020 [3] Gladieux, P., Feurtey, A., Hood, M. E., Snirc, A., Clavel, J., Dutech, C., Roy, M., & Giraud, T. (2015). The population biology of fungal invasions.Molecular Ecology, 24(9), 1969-86. https://doi.org/10.1111/mec.13028 [4] Fontyn, C., Zippert, A. C., Delestre, G., Marcel, T. C., Suffert, F., & Goyeau, H. (2022). Is virulence phenotype evolution driven exclusively by Lr gene deployment in French Puccinia triticina populations?. Plant Pathology, 71(7), 1511-1524. https://doi.org/10.1111/ppa.13599 [5] Fontyn, C., Meyer, K. J., Boixel, A. L., Delestre, G., Piaget, E., Picard, C., Suffer, F., Marcel, T.C., & Goyeau, H. (2022). Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations. bioRxiv, 2022.08.29.505401, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.29.505401 | Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations | Cécilia FONTYN, Kevin JG MEYER, Anne-Lise BOIXEL, Ghislain DELESTRE, Emma PIAGET, Corentin PICARD, Frédéric SUFFERT, Thierry C MARCEL, Henriette GOYEAU | <p style="text-align: justify;">Plant pathogens are constantly evolving and adapting to their environment, including their host. Virulence alleles emerge, and then increase, and sometimes decrease in frequency within pathogen populations in respon... | | Coevolution, Epidemiology, Evolution of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Pathogenic/Symbiotic Fungi, Phytopathology, Plant diseases, Population dynamics of hosts, infectious agents, or... | Pierre Gladieux | Emerson Del Ponte , Jacqui Shykoff, Leïla Bagny Beilhe , Alexey Mikaberidze | 2022-09-29 20:01:57 |




