
Latest recommendations

| Id | Title * ▲ | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
14 Feb 2024
A Bayesian analysis of birth pulse effects on the probability of detecting Ebola virus in fruit batsEpidemiological modeling to optimize the detection of zoonotic viruses in wild (reservoir) speciesRecommended by Aurelien Tellier based on reviews by Hetsron Legrace NYANDJO BAMEN and 1 anonymous reviewer based on reviews by Hetsron Legrace NYANDJO BAMEN and 1 anonymous reviewer
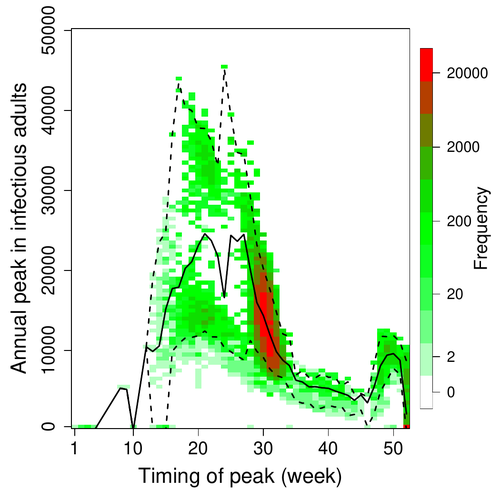
Various species of Ebolavirus have caused, and are still causing, zoonotic outbreaks and public health crises in Africa. Bats have long been hypothesized to be important reservoir populations for a series of viruses such as Hendra or Marburg viruses, the severe acute respiratory syndrome coronavirus (SARS-CoV, SARS-CoV-2) as well as Ebolaviruses [2, 3]. However the ecology of disease dynamics, disease transmission, and coevolution with their natural hosts of these viruses is still poorly understood, despite their importance for predicting novel outbreaks in human or livestock populations. The evidence that bats function as sylvatic reservoirs for Ebola viruses is yet only partial. Indeed, only few serological studies demonstrated the presence of Ebolavirus antibodies in young bats [4], albeit without providing positive controls of viral detection or identifying the viral species (via PCR). There is thus an unexplained discrepancy between serological data and viral detection [2, 4]. In this article, Pleydell et al. [1] use a modeling approach as well as published serological and age-structure (of the bat population) data to calibrate the model simulations. The study starts with the development of an age-structured epidemiological model which includes seasonal birth pulses and waning immunity, both generating pulses of Ebolavirus transmission within a colony of African straw-coloured fruit bats (Eidolon helvum). The epidemiological dynamics of such system of ordinary differential equations can generate annual outbreaks, skipped years or multi-annual cycles up to chaotic dynamics. Therefore, the calibration of the parameters, and the definition of biologically relevant priors, are key. To this aim, the serological data are obtained from a previous study in Cameroon [5], and the age structured of the bat population (birth and mortality) from a population study in Ghana [6]. These data are integrated into the Bayesian analysis and statistical framework to fit the model and generate predictions. In a nutshell, the authors show an overlap between the data and credibility intervals generated by the calibrated model, which thus explains well the seasonality of age-structure, namely changes in pup presence, number of lactating females, or proportion of juveniles in May. The authors can estimate that 76% of adults and 39% of young bats do survive each year, and infections are expected to last one and a half weeks. The epidemiological model predicts that annual birth pulses likely generate annual disease outbreaks, so that weeks 30 to 31 of each year, are predicted to be the best period to isolate the circulating Ebolavirus in this bat population. From the model predictions, the authors estimate the probability to have missed an infectious bat among all the samples tested by PCR being approximately of one per two thousands. The disease dynamics pattern observed in the serology data, and replicated by the model, is likely driven by seasonal pulses of young susceptible bats entering the population. This seasonal birth event increases the viral transmission, resulting in the observed peak of viral prevalence. With the inclusion of immunity waning and antibody persistence, the model results illuminate therefore why previous studies have detected only few positive cases by PCR tests, in contrast to the evidence from serological data. This study provides a first proof of principle that epidemiological modeling, despite its many simplifying assumptions, can be applied to wild species reservoirs of zoonotic diseases in order to optimize the design of field studies to detect viruses. Furthermore, such models can contribute to assess the probability and timing of zoonotic outbreaks in human or livestock populations. This article illustrates one of the manifold applications of mathematical theory of disease epidemiology to optimize sampling of pathogens/parasites or vaccine development and release [7, 8]. The further coupling of such models with population genetics theory and statistical inference methods (using parasite genome data) increasingly provide insights into the adaptation and evolution of parasites to human, crops and livestock populations [9, 10].
References [1] Pleydell D.R.J., Ndong Bass I., Mba Djondzo F.A., Djomsi D.M., Kouanfack C., Peeters M., and J. Cappelle. 2023. A Bayesian analysis of birth pulse effects on the probability of detecting Ebola virus in fruit bats. bioRxiv, ver. 3 peer reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2023.08.10.552777 [2] Caron A., Bourgarel M., Cappelle J., Liégeois F., De Nys H.M., and F. Roger. 2018. Ebola virus maintenance: if not (only) bats, what else? Viruses 10, 549. https://doi.org/10.3390/v10100549 [3] Letko M., Seifert S.N., Olival K.J., Plowright R.K., and V.J. Munster. 2020. Bat-borne virus diversity, spillover and emergence. Nature Reviews Microbiology 18, 461–471. https://doi.org/10.1038/s41579-020-0394-z [4] Leroy E.M., Kumulungui B., Pourrut X., Rouquet P., Hassanin A., Yaba P., Délicat A., Paweska J.T., Gonzalez J.P., and R. Swanepoel. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438, 575–576. https://doi.org/10.1038/438575a [5] Djomsi D.M. et al. 2022. Dynamics of antibodies to Ebolaviruses in an Eidolon helvum bat colony in Cameroon. Viruses 14, 560. https://doi.org/10.3390/v14030560 [6] Peel A.J. et al. 2016. Bat trait, genetic and pathogen data from large-scale investigations of African fruit bats Eidolon helvum. Scientific data 3, 1–11. https://doi.org/10.1038/sdata.2016.49 [7] Nyandjo Bamen H.L., Ntaganda J.M., Tellier A. and O. Menoukeu Pamen. 2023. Impact of imperfect vaccine, vaccine trade-off and population turnover on infectious disease dynamics. Mathematics, 11(5), p.1240. https://doi.org/10.3390/math11051240 [8] Saadi N., Chi Y.L., Ghosh S., Eggo R.M., McCarthy C.V., Quaife M., Dawa J., Jit M. and A. Vassall. 2021. Models of COVID-19 vaccine prioritisation: a systematic literature search and narrative review. BMC medicine, 19, pp.1-11. https://doi.org/10.1186/s12916-021-02190-3 [9] Maerkle, H., John S., Metzger, L., STOP-HCV Consortium, Ansari, M.A., Pedergnana, V. and Tellier, A., 2023. Inference of host-pathogen interaction matrices from genome-wide polymorphism data. bioRxiv, https://doi.org/10.1101/2023.07.06.547816. [10] Gandon S., Day T., Metcalf C.J.E. and B.T. Grenfell. 2016. Forecasting epidemiological and evolutionary dynamics of infectious diseases. Trends in ecology & evolution, 31(10), pp.776-788. https://doi.org/10.1016/j.tree.2016.07.010 | A Bayesian analysis of birth pulse effects on the probability of detecting Ebola virus in fruit bats | David R.J. Pleydell, Innocent Ndong Bass, Flaubert Auguste Mba Djondzo, Dowbiss Meta Djomsi, Charles Kouanfack, Martine Peeters, Julien Cappelle | <p>Since 1976 various species of Ebolavirus have caused a series of zoonotic outbreaks and public health crises in Africa. Bats have long been hypothesised to function as important hosts for ebolavirus maintenance, however the transmission ecology... | | Animal diseases, Disease Ecology/Evolution, Ecohealth, Ecology of hosts, infectious agents, or vectors, Epidemiology, Population dynamics of hosts, infectious agents, or vectors, Reservoirs, Viruses, Zoonoses | Aurelien Tellier | 2023-08-16 16:57:05 | ||
27 Feb 2023
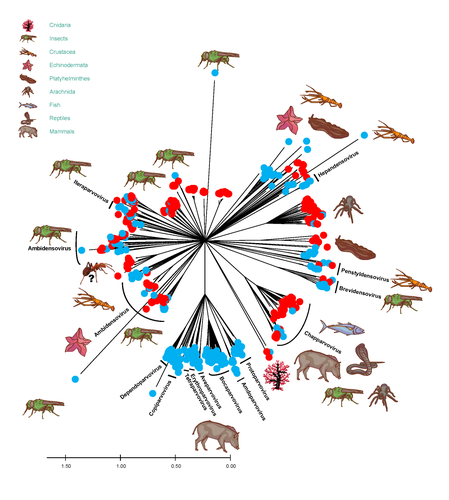
African army ants at the forefront of virome surveillance in a remote tropical forestA groundbreaking study using ants revealed a spectacular diversity of viruses in hardly accessible ecosystems like tropical forestsRecommended by Sebastien Massart based on reviews by Mart Krupovic and 1 anonymous reviewerDeciphering the virome (the set or assemblage of viruses) of the Earth, from individual organisms to entire ecosystems, has become a key priority. The first step to better understanding the impact of viruses on the ecology and functions of ecosystems is to describe their diversity. Such knowledge opens the gates to a better assessment of global nutrient cycling or of the threat that viruses represent to individual health. This explains the increasing number of pioneering studies that are currently sequencing the complete or partial genome of thousands of new viruses [1]. In their exciting study, Fritz and collaborators [2], authors sampled 209 army ants (Genus Dorylus) to investigate the virus diversity in dense forests that researchers cannot easily access. Indeed, these ants live in colonies (21 were sampled) that can move 1 km per day, covering a significant area and attacking many invertebrate and vertebrate preys. Each sample was sequenced by a protocol called VANA sequencing and allowing the enrichment of the sample in viral sequences [3], so improving the detection of viruses present at low abundance in the ant (and more specifically in its gut for viruses infecting preys). Around 45,000 contigs presented homologies with bacterial, plant, invertebrate, and vertebrate infecting viruses. Half could be assigned to 56 families and 157 genera of the International Committee on Taxonomy of Viruses. Beyond this amazing harvest of new and known virus sequences using an original methodology, the results significantly improve the current frontiers of known viral taxonomy and diversity and raise exciting research tracks to expand them. As a preprint, several blogs or news of leading scientists and journals have already highlighted this study. For example, in the news section of Science magazine, Jon Cohen underlined the originality of the approach for virus hunting on Earth with the title “Armed with air samplers, rope tricks, and—yes—ants, virus hunters spot threats in new ways”[4]. Another example is the mention of the publication by Elisabeth Bik in her Microbiome Digest: she wrote, “An amazing read is a fresh preprint from Fritz and collaborator describing an exciting method of sampling in difficult-to-reach environments“ [5]. The paper from Fritz et al [2] thus represents a significant advance in virus ecology, as already recognized by early readers, and this is why I strongly recommend its publication in PCI Infections. REFERENCES 1. Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, Lohr D, Novakovsky G, Buchfink B, Al-Shayeb B, Banfield JF, de la Peña M, Korobeynikov A, Chikhi R, Babaian A (2022) Petabase-scale sequence alignment catalyses viral discovery. Nature, 602, 142–147. https://doi.org/10.1038/s41586-021-04332-2 2. Fritz M, Reggiardo B, Filloux D, Claude L, Fernandez E, Mahé F, Kraberger S, Custer JM, Becquart P, Mebaley TN, Kombila LB, Lenguiya LH, Boundenga L, Mombo IM, Maganga GD, Niama FR, Koumba J-S, Ogliastro M, Yvon M, Martin DP, Blanc S, Varsani A, Leroy E, Roumagnac P (2023) African army ants at the forefront of virome surveillance in a remote tropical forest. bioRxiv, 2022.12.13.520061, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.12.13.520061 3. François S, Filloux D, Fernandez E, Ogliastro M, Roumagnac P (2018) Viral Metagenomics Approaches for High-Resolution Screening of Multiplexed Arthropod and Plant Viral Communities. In: Viral Metagenomics: Methods and Protocols Methods in Molecular Biology. (eds Pantaleo V, Chiumenti M), pp. 77–95. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7683-6_7 4. Cohen J (2023) Virus hunters test new surveillance tools. Science, 379, 16–17. https://doi.org/10.1126/science.adg5292 5. Ponsero A (2023) February 18th, 2023. Microbiome Digest - Bik’s Picks. https://microbiomedigest.com/2023/02/18/february-18th-2023/ | African army ants at the forefront of virome surveillance in a remote tropical forest | Matthieu Fritz, Berenice Reggiardo, Denis Filloux, Lisa Claude, Emmanuel Fernandez, Frederic Mahe, Simona Kraberger, Joy M. Custer, Pierre Becquart, Telstar Ndong Mebaley, Linda Bohou Kombila, Leadisaelle H. Lenguiya, Larson Boundenga, Illich M. M... | <p style="text-align: justify;">In this study, we used a predator-enabled metagenomics strategy to sample the virome of a remote and difficult-to-access densely forested African tropical region. Specifically, we focused our study on the use of arm... | | Ecohealth, Ecology of hosts, infectious agents, or vectors, One Health, Reservoirs, Viruses | Sebastien Massart | 2022-12-14 11:57:40 | ||
08 Aug 2023
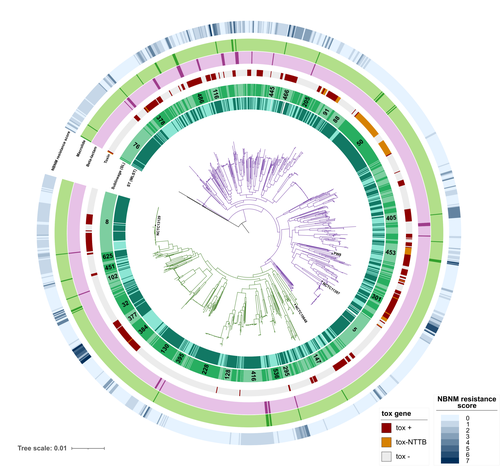
A global Corynebacterium diphtheriae genomic framework sheds light on current diphtheria reemergenceDIPHTOSCAN : A new tool for the genomic surveillance of diphtheriaRecommended by Rodolfo García-Contreras based on reviews by Ankur Mutreja and 2 anonymous reviewersOne of the greatest achievements of health sciences is the eradication of infectious diseases such as smallpox that in the past imposed a severe burden on humankind, through global vaccination campaigns. Moreover, progress towards the eradication of others such as poliomyelitis, dracunculiasis, and yaws is being made. In contrast, other infections that were previously contained are reemerging, due to several factors, including lack of access to vaccines due to geopolitical reasons, the rise of anti-vaccine movements, and the constant mobility of infected persons from the endemic sites. One of such disease is diphtheria, caused by Corynebacterium diphtheriae and a few other related species such as C. ulcerans and C. pseudotuberculosis. Importantly, in France, diphtheria cases reported in 2022 increased 7-fold from the average of previously recorded cases per year in the previous 4 years and the situation in other European countries is similar. Hence, as reported here, Hennart et al. (2023) developed DIPHTOSCAN, a free access bioinformatics tool with user-friendly interphase, aimed to easily identify, extract and interpret important genomic features such as the sublineage of the strain, the presence of the tox gene (as a string predictor for toxigenic disease) as well as genes coding other virulence factors such as fimbriae, and the presence of know resistant mechanisms towards antibiotics like penicillin and erythromycin currently used in the clinic to treat this infection. The authors validated the performance of their tool with a large collection of genomes, including those obtained from the isolates of the 2022 outbreak in France, more than 1,200 other genomes isolated from France, Algeria, and Yemen, and more than 500 genomes from several countries from Europe, America, Africa, Asia, and Oceania that are available through the NCBI site. DIPHTOSCAN will allow the rapid identification and surveillance of potentially dangerous strains such as those being tox-positive isolates and resistant to multiple drugs and/or first-line treatments and a better understanding of the epidemiology and evolution of this important reemerging disease. | A global *Corynebacterium diphtheriae* genomic framework sheds light on current diphtheria reemergence | Melanie Hennart, Chiara Crestani, Sebastien Bridel, Nathalie Armatys, Sylvie Brémont, Annick Carmi-Leroy, Annie Landier, Virginie Passet, Laure Fonteneau, Sophie Vaux, Julie Toubiana, Edgar Badell, Sylvain Brisse | <p style="text-align: justify;"><strong>Background</strong></p> <p style="text-align: justify;">Diphtheria, caused by <em>Corynebacterium diphtheriae</em>, reemerges in Europe since 2022. Genomic sequencing can inform on transmission routes and g... | | Drug resistance, tolerance and persistence, Epidemiology, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Microbiology of infections, Population genetics of hosts, infectiou... | Rodolfo García-Contreras | Ankur Mutreja | 2023-03-09 16:02:27 | |
19 Jul 2023
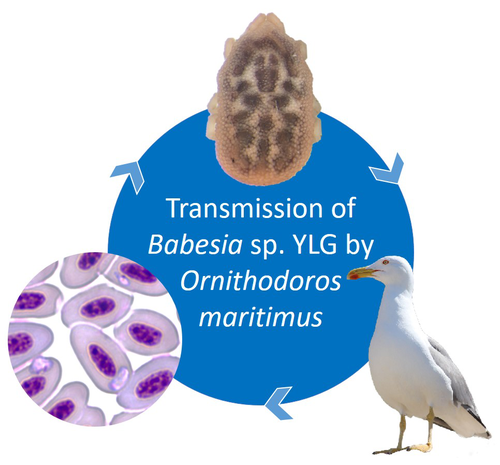
A soft tick vector of Babesia sp. YLG in Yellow-legged gull (Larus michahellis) nestsA four-year study reveals the potential role of the soft tick Ornithodoros maritimus in the transmission and circulation of Babesia sp. YLG in Yellow-legged gull colonies.Recommended by Thomas Pollet based on reviews by Hélène Jourdan-Pineau and Tahar KernifWorldwide, ticks and tick-borne diseases are a persistent example of problems at the One Health interface between humans, wildlife, and environment (1, 2). The management and prevention of ticks and tick-borne diseases require a better understanding of host, tick and pathogen interactions and thus get a better view of the tick-borne pathosystems. In this study (3), the tick-borne pathosystem included three component species: first a seabird host, the Yellow-legged gull (YLG - Larus michahellis, Laridae), second a soft nidicolous tick (Ornithodoros maritimus, Argasidae, syn. Alectorobius maritimus) known to infest this host and third a blood parasite (Babesia sp. YLG, Piroplasmidae). In this pathosystem, authors investigated the role of the soft tick, Ornithodoros maritimus, as a potential vector of Babesia sp. YLG. They analyzed the transmission of Babesia sp. YLG by collecting different tick life stages from YLG nests during 4 consecutive years on the islet of Carteau (Gulf of Fos, Camargue, France). Ticks were dissected and organs were analyzed separately to detect the presence of Babesia sp DNA and to evaluate different transmission pathways. While the authors detected Babesia sp. YLG DNA in the salivary glands of nymphs, females and males, this result reveals a strong suspicion of transmission of the parasite by the soft tick. Babesia sp. YLG DNA was also found in tick ovaries, which could indicate possible transovarial transmission. Finally, the authors detected Babesia sp. YLG DNA in several male testes and in endospermatophores, and notably in a parasite-free female (uninfected ovaries and salivary glands). These last results raise the interesting possibility of sexual transmission from infected males to uninfected females. As pointed out by both reviewers, this is a nice study, well written and easy to read. All the results are new and allow to better understand the role of the soft tick, Ornithodoros maritimus, as a potential vector of Babesia sp. YLG. They finally question about the degree to which the parasite can be maintained locally by ticks and the epidemiological consequences of infection for both O. maritimus and its avian host. For all these reasons, I chose to recommend this article for Peer Community In Infections. References
| A soft tick vector of *Babesia* sp. YLG in Yellow-legged gull (*Larus michahellis*) nests | Claire Bonsergent, Marion Vittecoq, Carole Leray, Maggy Jouglin, Marie Buysse, Karen D. McCoy, Laurence Malandrin | <p style="text-align: justify;"><em>Babesia </em>sp. YLG has recently been described in Yellow-legged gull (<em>Larus michahellis</em>) chicks and belongs to the Peircei clade in the new classification of Piroplasms. Here, we studied <em>Babesia <... | | Ecology of hosts, infectious agents, or vectors, Eukaryotic pathogens/symbionts, Interactions between hosts and infectious agents/vectors, Parasites, Vectors | Thomas Pollet | 2023-03-29 14:33:40 | ||
17 Jan 2024
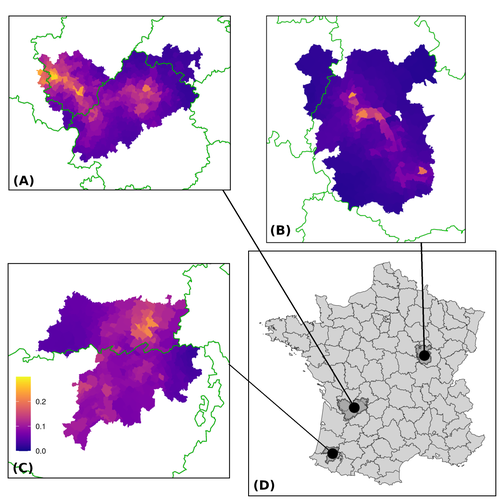
Assessing the dynamics of Mycobacterium bovis infection in three French badger populationsFrom disease surveillance to public action. Re-inforcing both epidemiological surveillance and data analysis: an illustration with Mycobacterium bovisRecommended by Jean-Francois Guégan based on reviews by Rowland Kao and 1 anonymous reviewerMycobacterium bovis, also called M. tuberculosis var. bovis, is a bacterium belonging to the M. tuberculosis complex (i.e., MTBC) and which can cause through zoonotic transmission another form of human tuberculosis (Tb). It is above all the agent of bovine tuberculosis (i.e., bTb) which affects not only cattle (wild or farmed) but also a large diversity of other wild mammals worldwide. An increasing number of infected animal cases are being discovered in many regions of the world, thus raising the problem of tuberculosis transmission, including to humans, more complex than previously thought. Efforts have been made in terms of vaccination or culling of populations of host carrier species, such as the badger for example, however leading to consequences of greater dispersion of the infectious agent. M. bovis shows a more or less significant capacity to persist outside its hosts, particularly in the environment under certain abiotic and biotic conditions. This bacillus can be transmitted and spread in many ways, including through aerosol, mucus and sputum, urine and feces, by direct contact with infected animals, their dead bodies or rather via their excreta or by inhalation of aerosols, depending on the host species concerned. In this paper, Calenge and his collaborators (Callenge et al. 2024) benefited from a national surveillance program on M. bovis cases in wild species, set up in 2011 in France, i.e., Sylvatub, for detecting and monitoring M. bovis infection in European badger (Meles meles) populations. Sylvatub is a participatory program involving both national and local stakeholder systems in order to determine changes in bTb infection levels in domestic and wild animal species. This original work had two aims: to describe spatial disease dynamics in the three clusters under scrutiny using a complex Bayesian model; and to develop indicators for the monitoring of the M. bovis infection by stakeholders and decision-makers of the program. This paper is timely and very comprehensive. In this cogent study, the authors illustrate this point by using epidemiological surveillance to obtain large amounts of data (which is generally lacking in human epidemiology, but more dramatically lacking in animal epidemiology) and a highly sophisticated biostatistical analysis (Callenge et al. 2024). It is in itself a demonstration of the current capabilities of population dynamics applied to infectious disease situations, in this case animal, in the rapidly developing discipline of disease ecology and evolution. One of the aims of the study is to propose statistical models that can be used by the different stakeholders in charge, for instance, of wildlife conservation or the regional or State veterinary services to assess disease risk in the most affected regions. References Assel AKHMETOVA, Jimena GUERRERO, Paul McADAM, Liliana CM SALVADOR, Joseph CRISPELL, John LAVERY, Eleanor PRESHO, Rowland R KAO, Roman BIEK, Fraser MENZIES, Nigel TRIMBLE, Roland HARWOOD, P Theo PEPLER, Katarina ORAVCOVA, Jordon GRAHAM, Robin SKUCE, Louis DU PLESSIS, Suzan THOMPSON, Lorraine WRIGHT, Andrew W BYRNE, Adrian R ALLEN. 2023. Genomic epidemiology of Mycobacterium bovis infection in sympatric badger and cattle populations in Northern Ireland. Microbial Genomics 9: mgen001023. https://doi.org/10.1099/mgen.0.001023 Roman BIEK, Anthony O’HARE, David WRIGHT, Tom MALLON, Carl McCORMICK, Richard J ORTON, Stanley McDOWELL, Hannah TREWBY, Robin A SKUCE, Rowland R KAO. 2012. Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathogens 8: e1003008. https://doi.org/10.1371/journal.ppat.1003008 Clément CALENGE, Ariane PAYNE, Edouard REVEILLAUD, Céline RICHOMME, Sébastien GIRARD, Stephanie DESVAUX. 2024. Assessing the dynamics of Mycobacterium bovis infection in three French badger populations. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2023.05.31.543041 Marc CHOISY, Pejman ROHANI. 2006. Harvesting can increase severity of wildlife disease epidemics. Proceedings of the Royal Society, London, Ser. B 273: 2025-2034. https://doi.org/10.1098/rspb.2006.3554 Shannon C DUFFY, Sreenidhi SRINIVASAN, Megan A SCHILLING, Tod STUBER, Sarah N DANCHUK, Joy S MICHAEL, Manigandan VENKATESAN, Nitish BANSAL, Sushila MAAN, Naresh JINDAL, Deepika CHAUDHARY, Premanshu DANDAPAT, Robab KATANI, Shubhada CHOTHE, Maroudam VEERASAMI, Suelee ROBBE-AUSTERMAN, Nicholas JULEFF, Vivek KAPUR, Marcel A BEHR. 2020. Reconsidering Mycobacterium bovis as a proxy for zoonotic tuberculosis: a molecular epidemiological surveillance study. Lancet Microbe 1: e66-e73. https://doi.org/10.1016/S2666-5247(20)30038-0 Jean-François GUEGAN. 2019. The nature of ecology of infectious disease. The Lancet Infectious Diseases 19. https://doi.org/10.1016/s1473-3099(19)30529-8 Brandon H HAYES, Timothée VERGNE, Mathieu ANDRAUD, Nicolas ROSE. 2023. Mathematical modeling at the livestock-wildlife interface: scoping review of drivers of disease transmission between species. Frontiers in Veterinary Science 10: 1225446. https://doi.org/10.3389/fvets.2023.1225446 David KING, Tim ROPER, Douglas YOUNG, Mark EJ WOOLHOUSE, Dan COLLINS, Paul WOOD. 2007. Bovine tuberculosis in cattle and badgers. Report to Secretary of State about tuberculosis in cattle and badgers. London, UK. https://www.bovinetb.info/docs/RBCT_david_%20king_report.pdf Robert MM SMITH , Francis DROBNIEWSKI, Andrea GIBSON, John DE MONTAGUE, Margaret N LOGAN, David HUNT, Glyn HEWINSON, Roland L SALMON, Brian O’NEILL. 2004. Mycobacterium bovis Infection, United Kingdom. Emerging Infectious Diseases 10: 539-541. https://doi.org/10.3201/eid1003.020819 | Assessing the dynamics of *Mycobacterium bovis* infection in three French badger populations | Clement CALENGE, Ariane PAYNE, Edouard REVEILLAUD, Celine RICHOMME, Sebastien GIRARD, Stephanie DESVAUX | <p>The Sylvatub system is a national surveillance program established in 2011 in France to monitor infections caused by <em>Mycobacterium bovis</em>, the main etiologic agent of bovine tuberculosis, in wild species. This participatory program, inv... | | Animal diseases, Ecohealth, Ecology of hosts, infectious agents, or vectors, Epidemiology, Geography of infectious diseases, Pathogenic/Symbiotic Bacteria, Zoonoses | Jean-Francois Guégan | 2023-06-05 10:50:49 | ||
29 Jan 2024
Celebrating the 20th Anniversary of the First Xanthomonas Genome Sequences – How Genomics Revolutionized Taxonomy, Provided Insight into the Emergence of Pathogenic Bacteria, Enabled New Fundamental Discoveries and Helped Developing Novel Control Measures – A Perspective from the French Network on XanthomonadsAdvancing Pathogen Genomics: A Comprehensive Review of the Xanthomonas(*) Genome's Impact on Bacterial Research and Control StrategiesRecommended by Damien François Meyer based on reviews by Boris Vinatzer and 3 anonymous reviewers
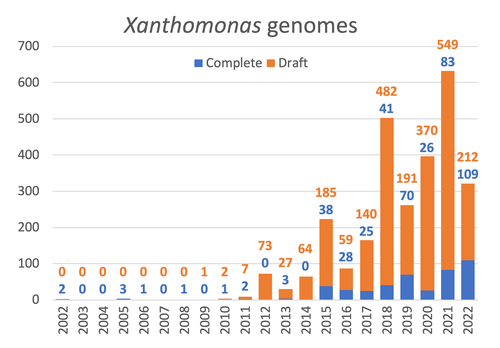
The paper titled "Celebrating the 20th Anniversary of the First Xanthomonas Genome Sequences – How Genomics Revolutionized Taxonomy Provided Insight into the Emergence of Pathogenic Bacteria Enabled New Fundamental Discoveries and Helped Developing Novel Control Measures – A Perspective from the French Network on Xanthomonads" by Ralf Koebnik et al. (2023) is an insightful contribution to the field of genomics and its application in understanding pathogenic bacteria, particularly Xanthomonas. This comprehensive review offers a unique perspective from the French Network on Xanthomonads, underscoring the significant advancements in taxonomy, pathogen emergence, and development of control strategies due to genomic research. One of the paper's main strengths is its thorough exploration of how genomics has revolutionized our understanding of Xanthomonas and other pathogenic bacteria. It sheds light on the evolution and emergence of these pathogens, contributing significantly to the development of novel and effective control measures. The authors' detailed account of the historical progress and current state of genomics in this field highlights its pivotal role in guiding future research and practical applications in managing bacterial diseases. Moreover, the paper emphasizes the importance of collaborative efforts and the sharing of knowledge within scientific networks, as exemplified by the French Network on Xanthomonas. This approach not only enriches the study but also serves as a model for future collaborative research endeavors. In conclusion, the work of Koebnik et al. is a valuable resource for researchers, policymakers, and practitioners in the field of plant pathology and genomics. It not only provides a comprehensive overview of the advances in genomics related to Xanthomonas but also illustrates the broader impact of genomic studies in understanding and managing pathogenic bacteria. References Ralf Koebnik, Sophie Cesbron, Nicolas W. G. Chen, Marion Fischer-Le Saux, Mathilde Hutin, Marie-Agnès Jacques, Laurent D. Noël, Alvaro Perez-Quintero, Perrine Portier, Olivier Pruvost, Adrien Rieux, And Boris Szurek (2024) Celebrating the 20th anniversary of the first Xanthomonas genome gequences – How genomics revolutionized taxonomy, provided insight into the emergence of pathogenic bacteria, enabled new fundamental discoveries and helped developing novel control measures – A perspective from the French network on Xanthomonads. Zenodo ver. 3, peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.8223857 | Celebrating the 20th Anniversary of the First Xanthomonas Genome Sequences – How Genomics Revolutionized Taxonomy, Provided Insight into the Emergence of Pathogenic Bacteria, Enabled New Fundamental Discoveries and Helped Developing Novel Control ... | Ralf Koebnik, Sophie Cesbron, Nicolas W. G. Chen, Marion Fischer-Le Saux, Mathilde Hutin, Marie-Agnès Jacques, Laurent D. Noël, Alvaro Perez-Quintero, Perrine Portier, Olivier Pruvost, Adrien Rieux, And Boris Szurek | <p>In this Opinion paper, members of the French Network on Xanthomonads give their personal view on what they consider to be some of the groundbreaking discoveries in the field of molecular plant pathology over the past 20 years. By celebrating th... | | Epidemiology, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Molecular biology of infections, Molecular genetics o... | Damien François Meyer | 2023-08-09 10:37:15 | ||
21 Sep 2023
Chikungunya intra-vector dynamics in Aedes albopictus from Lyon (France) upon exposure to a human viremia-like dose range reveals vector barrier permissiveness and supports local epidemic potentialFill in one gap in our understanding of CHIKV intra-vector dynamicsRecommended by Sara Moutailler based on reviews by 2 anonymous reviewers
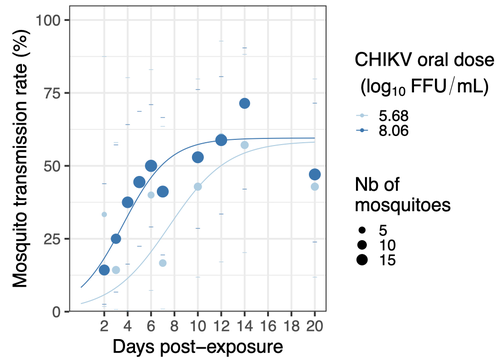
Mosquitoes are first vector of pathogen worldwide and transmit several arbovirus, most of them leading to major outbreaks (1). Chikungunya virus (CHIKV) is a perfect example of the “explosive type” of arbovirus, as observed in La Réunion Island in 2005-2006 (2-6) and also in the outbreak of 2007 in Italy (7), both vectorized by Ae. albopictus. Being able to better understand CHIKV intra-vector dynamics is still of major interest since not all chikungunya strain are explosive ones (8). In this study (9), the authors have evaluated the vector competence of a local strain of Aedes albopictus (collected in Lyon, France) for CHIKV. They evaluated infection, dissemination and transmission dynamics of CHIKV using different dose of virus in individual mosquitoes from day 2 to day 20 post exposure, by titration and quantification of CHIKV RNA load in the saliva. As highlighted by both reviewers, the most innovative idea in this study was the use of three different oral doses trying to span human viraemia detected in two published studies (10-11), doses that were estimated through their model of human CHIKV viremia in the blood. They have found that CHIKV dissemination from the Ae. albopictus midgut depends on the interaction between time post-exposure and virus dose (already highlighted by other international publications). Then their results were implemented in the agent-based model nosoi to estimate the epidemic potential of CHIKV in a French population of Ae. albopictus, using realistic vectorial capacity parameters. To conclude, the authors have discussed the importance of other parameters that could influence vector competence as mosquito microbiota and temperature, parameters that need also to be estimated in local mosquito population to improve the risk assessment through modelling. As pointed out by both reviewers, this is a nice study, well written and easy to read. These results allow filling in another gap of our understanding of CHIKV intra-vector dynamics and highlight the epidemic potential of CHIKV upon transmission by Aedes albopictus in mainland France. For all these reasons, I chose to recommend this article for Peer Community In Infections. References 1. Marine Viglietta, Rachel Bellone, Adrien Albert Blisnick, Anna-Bella Failloux. (2021). Vector Specificity of Arbovirus Transmission. Front Microbiol Dec 9;12:773211. https://doi.org/10.3389/fmicb.2021.773211 2. Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney M-C, Lavenir R, Pardigon N, Reynes J-M, Pettinelli F, Biscornet L, Diancourt L, Michel S, Duquerroy S, Guigon G, Frenkiel M-P, Bréhin A-C, Cubito N, Desprès P, Kunst F, Rey FA, Zeller H, Brisse S. (2006). Genome Microevolution of Chikungunya viruses Causing the Indian Ocean Outbreak. 2006. PLoS Medicine, 3, e263. https://doi.org/10.1371/journal.pmed.0030263 3. Bonilauri P, Bellini R, Calzolari M, Angelini R, Venturi L, Fallacara F, Cordioli P, 687 Angelini P, Venturelli C, Merialdi G, Dottori M. (2008). Chikungunya Virus in Aedes albopictus, Italy. Emerging Infectious 689 Diseases, 14, 852–854. https://doi.org/10.3201/eid1405.071144 4. Pagès F, Peyrefitte CN, Mve MT, Jarjaval F, Brisse S, Iteman I, Gravier P, Tolou H, Nkoghe D, Grandadam M. (2009). Aedes albopictus Mosquito: The Main Vector of the 2007 Chikungunya Outbreak in Gabon. PLoS ONE, 4, e4691. https://doi.org/10.1371/journal.pone.0004691 5. Paupy C, Kassa FK, Caron M, Nkoghé D, Leroy EM (2012) A Chikungunya Outbreak Associated with the Vector Aedes albopictus in Remote Villages of Gabon. Vector-Borne and Zoonotic Diseases, 12, 167–169. https://doi.org/10.1089/vbz.2011.0736 6. Mombouli J-V, Bitsindou P, Elion DOA, Grolla A, Feldmann H, Niama FR, Parra H-J, Munster VJ. (2013). Chikungunya Virus Infection, Brazzaville, Republic of Congo, 2011. Emerging Infectious Diseases, 19, 1542–1543. https://doi.org/10.3201/eid1909.130451 7. Venturi G, Luca MD, Fortuna C, Remoli ME, Riccardo F, Severini F, Toma L, Manso MD, Benedetti E, Caporali MG, Amendola A, Fiorentini C, Liberato CD, Giammattei R, Romi R, Pezzotti P, Rezza G, Rizzo C. (2017). Detection of a chikungunya outbreak in Central Italy, August to September 2017. Eurosurveillance, 22, 17–00646. https://doi.org/10.2807/1560-7917.es.2017.22.39.17-00646 8. de Lima Cavalcanti, T.Y.V.; Pereira, M.R.; de Paula, S.O.; Franca, R.F.d.O. (2022). A Review on Chikungunya Virus Epidemiology, Pathogenesis and Current Vaccine Development. Viruses 2022, 14, 969. https://doi.org/10.3390/v14050969 9. Barbara Viginier, Lucie Cappuccio, Celine Garnier, Edwige Martin, Carine Maisse, Claire Valiente Moro, Guillaume Minard, Albin Fontaine, Sebastian Lequime, Maxime Ratinier, Frederick Arnaud, Vincent Raquin. (2023). Chikungunya intra-vector dynamics in Aedes albopictus from Lyon (France) upon exposure to a human viremia-like dose range reveals vector barrier permissiveness and supports local epidemic potential. medRxiv, ver.3, peer-reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2022.11.06.22281997 10. Appassakij H, Khuntikij P, Kemapunmanus M, Wutthanarungsan R, Silpapojakul K (2013) Viremic profiles in CHIKV-infected cases. Transfusion, 53, 2567–2574. https://doi.org/10.1111/j.1537-2995.2012.03960.x 11. Riswari SF, Ma’roef CN, Djauhari H, Kosasih H, Perkasa A, Yudhaputri FA, Artika IM, Williams M, Ven A van der, Myint KS, Alisjahbana B, Ledermann JP, Powers AM, Jaya UA (2015) Study of viremic profile in febrile specimens of chikungunya in Bandung, Indonesia. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology, 74, 61–5. https://doi.org/10.1016/j.jcv.2015.11.017 | Chikungunya intra-vector dynamics in *Aedes albopictus* from Lyon (France) upon exposure to a human viremia-like dose range reveals vector barrier permissiveness and supports local epidemic potential | Barbara Viginier, Lucie Cappuccio, Celine Garnier, Edwige Martin, Carine Maisse, Claire Valiente Moro, Guillaume Minard, Albin Fontaine, Sebastian Lequime, Maxime Ratinier, Frederick Arnaud, Vincent Raquin | <p>Arbovirus emergence and epidemic potential, as approximated by the vectorial capacity formula, depends on host and vector parameters, including the vector intrinsic ability to replicate then transmit the pathogen known as vector competence. Vec... | | Epidemiology, Vectors, Viruses | Sara Moutailler | 2023-06-17 15:59:17 | ||
28 Oct 2022

Development of nine microsatellite loci for Trypanosoma lewisi, a potential human pathogen in Western Africa and South-East Asia, and preliminary population genetics analysesPreliminary population genetic analysis of Trypanosoma lewisiRecommended by Annette MacLeod based on reviews by Gabriele Schönian and 1 anonymous reviewerTrypanosoma lewisi is an atypical trypanosome species. Transmitted by fleas, it has a high prevalence and worldwide distribution in small mammals, especially rats [1]. Although not typically thought to infect humans, there has been a number of reports of human infections by T. lewisi in Asia including a case of a fatal infection in an infant [2]. The fact that the parasite is resistant to lysis by normal human serum [3] suggests that many people, especially immunocompromised individuals, may be at risk from zoonotic infections by this pathogen, particularly in regions where there is close contact with T. lewisi-infected rat fleas. Indeed, it is also possible that cryptic T. lewisi infections exist but have hitherto gone undetected. Such asymptomatic infections have been detected for a number of parasitic infections including the related parasite T. b. gambiense [4]. References
[2] Truc P, Büscher P, Cuny G, Gonzatti MI, Jannin J, Joshi P, Juyal P, Lun Z-R, Mattioli R, Pays E, Simarro PP, Teixeira MMG, Touratier L, Vincendeau P, Desquesnes M (2013) Atypical Human Infections by Animal Trypanosomes. PLOS Neglected Tropical Diseases, 7, e2256. https://doi.org/10.1371/journal.pntd.0002256 [3] Lun Z-R, Wen Y-Z, Uzureau P, Lecordier L, Lai D-H, Lan Y-G, Desquesnes M, Geng G-Q, Yang T-B, Zhou W-L, Jannin JG, Simarro PP, Truc P, Vincendeau P, Pays E (2015) Resistance to normal human serum reveals Trypanosoma lewisi as an underestimated human pathogen. Molecular and Biochemical Parasitology, 199, 58–61. https://doi.org/10.1016/j.molbiopara.2015.03.007 [4] Büscher P, Bart J-M, Boelaert M, Bucheton B, Cecchi G, Chitnis N, Courtin D, Figueiredo LM, Franco J-R, Grébaut P, Hasker E, Ilboudo H, Jamonneau V, Koffi M, Lejon V, MacLeod A, Masumu J, Matovu E, Mattioli R, Noyes H, Picado A, Rock KS, Rotureau B, Simo G, Thévenon S, Trindade S, Truc P, Reet NV (2018) Do Cryptic Reservoirs Threaten Gambiense-Sleeping Sickness Elimination? Trends in Parasitology, 34, 197–207. https://doi.org/10.1016/j.pt.2017.11.008 [5] Ségard A, Roméro A, Ravel S, Truc P, Gauthier D, Gauthier P, Dossou H-J, Sylvestre B, Houéménou G, Morand S, Chaisiri K, Noûs C, De Meeûs T (2022) Development of nine microsatellite loci for Trypanosoma lewisi, a potential human pathogen in Western Africa and South-East Asia, and preliminary population genetics analyses. Zenodo, 6460010, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.6460010 | Development of nine microsatellite loci for Trypanosoma lewisi, a potential human pathogen in Western Africa and South-East Asia, and preliminary population genetics analyses | Adeline Ségard, Audrey Romero, Sophie Ravel, Philippe Truc, Gauthier Dobigny, Philippe Gauthier, Jonas Etougbetche, Henri-Joel Dossou, Sylvestre Badou, Gualbert Houéménou, Serge Morand, Kittipong Chaisiri, Camille Noûs, Thierry deMeeûs | <p><em>Trypanosoma lewisi</em> belongs to the so-called atypical trypanosomes that occasionally affect humans. It shares the same hosts and flea vector of other medically relevant pathogenic agents as Yersinia pestis, the agent of plague. Increasi... | | Animal diseases, Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Eukaryotic pathogens/symbionts, Evolution of hosts, infectious agents, or vectors, Microbiology of infections, Parasites, Population genetics of hosts, in... | Annette MacLeod | 2022-04-21 17:04:37 | ||
24 Jun 2024
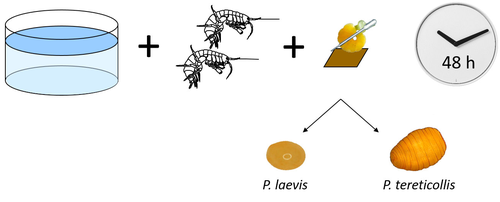
Differences in specificity, development time and virulence between two acanthocephalan parasites, infecting two cryptic species of Gammarus fossarumGammarid is not equal gammarid for acanthocephalan parasitesRecommended by Daniel Grabner based on reviews by 2 anonymous reviewersThe question on the role of different alternative hosts in the life cycle of acanthocephalan parasites has not been fully resolved to date. There is some information on the use of fish hosts in the genus Pomphorhynchus (Perrot-Minnot et al. 2019). It is known that acanthocephalans of the genus Pomphorhynchus can infect a number of different amphipod species (e.g. Bauer et al. 2000; Cornet et al. 2010; Dezfuli et al. 1999) but it is not clear if some host species might be more “advantageous” for the parasite, or if the parasite is more virulent to some host species than to others. Bauer et al. (2024) investigated different well characterized cryptic lineages of Gammarus fossarum (Weiss et al. 2013) for their susceptibility for two Pomphorhynchus sp. The results show that there is a difference in susceptibility to acanthocephalans between different linages of G. fossarum. Additionally, a parasite species specific difference was detected: the difference in susceptibility was more pronounced for P. tereticollis than for P. laevis. P. tereticollis was less virulent and developed slower than P. laevis (in G. fossarum). Besides the improved understanding of the biology of acanthocephalan parasites, this study clearly points out that we have to be careful with putting the “generalist” label on parasites simply due to the number of alternative host species we find them in. Instead, we should always have in mind that some of these hosts might be less suitable for the parasite than others when comparing quantitative data on the infection success. I highly appreciate the experimental approach taken that allows more profound conclusions than evaluations of field data. Experiments and analyses have been conducted well. I think this paper is significantly enhancing our knowledge on the specificity for the intermediate host. I find it highly remarkable that this was even found among different host lineages. References Bauer, A., Trouve, S., Gregoire, A., Bollache, L., Cezilly, F. (2000) Differential influence of Pomphorhynchus laevis (Acanthocephala) on the behaviour of native and invader gammarid species. International Journal for Parasitology, 30(14), 1453-1457. https://doi.org/10.1016/s0020-7519(00)00138-7 Bauer, A., Develay Nguyen, L., Motreuil, S., Teixeira, M., Debrosse, N., Rigaud, T. (2024) Experimental infections reveal differences in specificity, development time and virulence between the acanthocephalan parasite Pomphorhynchus tereticollis and its sympatric counterpart P. laevis, in two cryptic species of Gammarus fossarum. HAL, Ver. 2, Peer-Reviewed and Recommended by Peer Community in Infections, hal-04455823. https://hal.science/hal-04455823 Cornet, S., Sorci, G., Moret, Y. (2010) Biological invasion and parasitism: invaders do not suffer from physiological alterations of the acanthocephalan Pomphorhynchus laevis. Parasitology, 137(1), 137-147. https://doi.org/10.1017/S0031182009991077 Dezfuli, B.S., Rossetti, E., Bellettato, C.M., Maynard, B.J. (1999) Pomphorhynchus laevis in its intermediate host Echinogammarus stammeri in the River Brenta, Italy. Journal of Helminthology, 73(2), 95-102. https://doi.org/10.1017/S0022149X00700277 Perrot-Minnot, M.J., Guyonnet, E., Bollache, L., Lagrue, C. (2019) Differential patterns of definitive host use by two fish acanthocephalans occurring in sympatry: Pomphorhynchus laevis and Pomphorhynchus tereticollis. International Journal for Parasitology: Parasites and Wildlife, 8, 135-144. https://doi.org/10.1016/j.ijppaw.2019.01.007 Weiss, M., Macher, J.N., Seefeldt, M.A., Leese, F. (2013) Molecular evidence for further overlooked species within the Gammarus fossarum complex (Crustacea: Amphipoda). Hydrobiologia, 721(1), 165-184. https://doi.org/10.1007/s10750-013-1658-7
| Differences in specificity, development time and virulence between two acanthocephalan parasites, infecting two cryptic species of *Gammarus fossarum* | Alexandre Bauer, Lucie Develay Nguyen, Sébastien Motreuil, Maria Teixeira, Nelly Debrosse, Thierry Rigaud | <p style="text-align: justify;">Multi-host parasites can exploit various host species that differ in abundance and susceptibility to infection, which will contribute unequally to their transmission and fitness. Several species of acanthocephalan m... | | Ecology of hosts, infectious agents, or vectors, Evolution of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Molecular genetics of hosts, infectious agents, or vectors, Parasites, Resistance/Virulen... | Daniel Grabner | 2024-02-14 13:39:19 | ||
16 Jul 2024
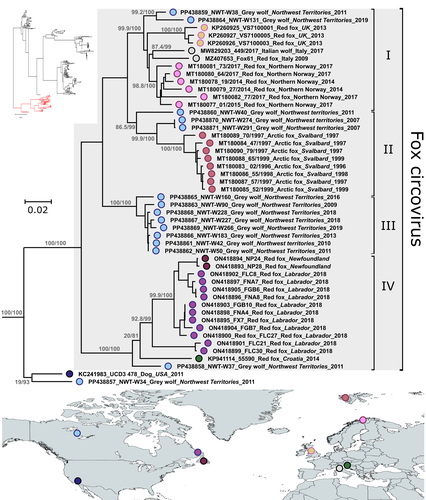
Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, CanadaWild canine viruses in the news. Better understanding multi-host transmission by adopting a disease ecology species community-based approachRecommended by Jean-Francois Guégan based on reviews by Arvind Varsani and 1 anonymous reviewerAccording to the international animal health authority, i.e., the World Organization on Animal Health (WOAH, former OIE), circoviruses are part of the Circoviridae family, which only includes 2 genera Circovirus and Cyclovirus, and infect swine, canine, ursid, viverrid, felid, pinniped, herpestid, mustelid, and several avian species (WOAH 2021). They are small (12–27 nm), non-enveloped, circular, single-stranded DNA viruses, viral replication is nuclear, and wild and domestic birds and mammals could serve as natural hosts. If most infections caused by circoviruses are subclinical in both wild and domestic species, they can be responsible for severe diseases in the commercial pig industry due to the Porcine circovirus-2 (PCV-2). These viruses can constitute a threat to wildlife, and cause their hosts to become immunocompromised, and animals often present with secondary coinfections. Canine circoviruses (CanineCV) harbour a worldwide distribution in dogs, and is the sole member of the viral genus to infect canines. They can be detected in wild carnivores, such as wolves, badgers, foxes and jackals, which indicates an ability for cross-species transmission between wildlife and domestic dogs. However, fox circovirus (FoCV), a distinct lineage of CanineCV, has been identified exclusively in wild canids (foxes and wolves) and not in dogs in Europe and North America, where it can cause in red foxes meningoencephalitis and other central nervous system signs. In their article, Canuti et al. (2024) investigate the presence, distribution and ecology of CanineCV in grey wolf specimens from the Northwest Territories, Canada. CanineCV occurrence appears to be relatively high with 45.3% positive specimens and parvoviral superinfections observed. The authors identify a high CanineCV genetic diversity among the investigated grey wolf specimens, and exacerbated by viral recombination. Phylogenetic analysis reveals the existence of 4 lineages, within each of them strains segregate by geography and not by host origin. This observed geographic segregation is interpreted as being due to the absence of exchange flows between grey wolf host subpopulations. Due to the paucity of knowledge on these circoviruses in wildlife and at the interface between wild and domestic animals, the authors discuss the plausible role of wolves as natural host reservoirs for disease transmission due to long-lasting virus-host coevolution. They are also conscious that additional maintenance hosts could exist in the wild, claiming for further studies to decipher fox circovirus disease ecology and transmission dynamics. This study underlines the importance of better understanding the transmission ecology and evolution of these Canine circoviruses, and I can only agree. Xiao et al. (2023), a research not referred to in the present work, evidenced CanineCV infection in cats in China, and obtained the first whole genome of cat-derived CanineCV. This emphasizes the importance of monitoring additional animal species and locations in the world to clarify disease ecology and transmission dynamics. A broader sampling of a wide range of animal species in different parts of the world using a species community-based approach is the key to understanding these CanineCV infections. References Marta CANUTI, Abigail V.L. KING, Giovanni FRANZO, H. Dean CLUFF, Lars E. LARSEN, Heather FENTON, Suzanne C. DUFOUR, Andrew S. LANG. 2024. Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, Canada. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2024.03.08.584028 World Organization on Animal Health. 2021. Circoviruses. https://www.woah.org/app/uploads/2021/05/circoviruses-infection-with.pdf [consulted on July 9th, 2024]. Xiangyu XIAO, Yan CHAO LI, Feng PEI XU, Xiangpi HAO, Shoujun LI, Pei ZHOU. 2023. Canine circovirus among dogs and cats in China: first identification in cats. Front. Microbiol. 14. https://doi.org/10.3389/fmicb.2023.1252272 | Diverse fox circovirus (*Circovirus canine*) variants circulate at high prevalence in grey wolves (*Canis lupus*) from the Northwest Territories, Canada | Marta Canuti, Abigail V.L. King, Giovanni Franzo, H. Dean Cluff, Lars E. Larsen, Heather Fenton, Suzanne C. Dufour, Andrew S. Lang | <p style="text-align: justify;">Canine circoviruses (CanineCV) have a worldwide distribution in dogs and are occasionally detected in wild carnivorans, indicating their ability for cross-species transmission. However, fox circovirus, a lineage of ... | | Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Epidemiology, Molecular genetics of hosts, infectious agents, or vectors, Population genetics of hosts, infectious agents, or vectors, Reservoirs, Taxonomy of hosts, infec... | Jean-Francois Guégan | Martine Peeters, Arvind Varsani | 2024-03-09 09:04:29 |




