
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * ▲ | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
25 Apr 2023

The distribution, phenology, host range and pathogen prevalence of Ixodes ricinus in France: a systematic map and narrative reviewAn extensive review of Ixodes ricinus in European FranceRecommended by Ana Sofia Santos based on reviews by Ana Palomar and 1 anonymous reviewerTicks are obligate, bloodsucking, nonpermanent ectoparasitic arthropods. Among them, Ixodes ricinus is a classic example of an extreme generalist tick, presenting a highly permissive feeding behavior using different groups of vertebrates as hosts, such as mammalian (including humans), avian and reptilian species (Hoogstraal & Aeschlimann, 1982; Dantas-Torresa & Otranto, 2013). This ecological adaptation can account for the broad geographical distribution of I. ricinus populations, which extends from the western end of the European continent to the Ural Mountains in Russia, and from northern Norway to the Mediterranean basin, including the North African countries - Morocco, Algeria and Tunisia (https://ecdc.europa.eu/en/disease-vectors/surveillance-and-disease-data/tick-maps). The contact with different hosts also promotes the exposure/acquisition and transmission of various pathogenic agents (viruses, bacteriae, protists and nematodes) of veterinary and medical relevance (Aeschlimann et al., 1979). As one of the prime ticks found on humans, this species is implicated in diseases such as Lyme borreliosis, Spotted Fever Group rickettsiosis, Human Anaplasmosis, Human Babesiosis and Tick-borne Encephalitis (Velez et al., 2023). The climate change projections drawn for I. ricinus, in the scenario of global warming, point for the expansion/increase activity in both latitude and altitude (Medlock et al., 2013). The adequacy of vector modeling is relaying in the proper characterization of complex biological systems. Thus, it is essential to increase knowledge on I. ricinus, focusing on aspects such as genetic background, ecology and eco-epidemiology on a microscale but also at a country and region level, due to possible local adaptations of tick populations and genetic drift. In the present systematic revision, Perez et al. (2023) combine old and recently published data (mostly up to 2020) regarding I. ricinus distribution, phenology, host range and pathogen association in continental France and Corsica Island. Based on a keyword search of peer-reviewed papers on seven databases, as well as other sources of grey literature (mostly, thesis), the authors have synthesized information on: 1) Host parasitism to detect potential differences in host use comparing to other areas in Europe; 2) The spatiotemporal distribution of I. ricinus, to identify possible geographic trends in tick density, variation in activity patterns and the influence of environmental factors; 3) Tick-borne pathogens detected in this species, to better assess their spatial distribution and variation in exposure risk. As pointed out by both reviewers, this work clearly summarizes the information regarding I. ricinus and associated microorganisms from European France. This review also identifies remaining knowledge gaps, providing a comparable basis to orient future research. This is why I chose to recommend Perez et al (2023)'s preprint for Peer Community Infections. REFERENCES Aeschlimann, A., Burgdorfer, W., Matile, H., Peter, O., Wyler, R. (1979) Aspects nouveaux du rôle de vecteur joué par Ixodes ricinus L. en Suisse. Acta Tropica, 36, 181-191. Dantas-Torresa, F., Otranto, D. (2013) Seasonal dynamics of Ixodes ricinus on ground level and higher vegetation in a preserved wooded area in southern Europe. Veterinary Parasitology, 192, 253- 258. Hoogstraal, H., Aeschlimann, A. (1982) Tick-host specificity. Mitteilungen der Schweizerischen Entomologischen Gesellschaft, 55, 5-32. Medlock, J.M., Hansford, K.M., Bormane, A., Derdakova, M., Estrada-Peña, A., George, J.C., Golovljova, I., Jaenson, T.G.T., Jensen, J.K., Jensen, P.M., Kazimirova, M., Oteo, J.A., Papa, A., Pfister, K., Plantard, O., Randolph, S.E., Rizzoli, A., Santos-Silva, M.M., Sprong, H., Vial, L., Hendrickx, G., Zeller, H., Van Bortel, W. (2013) Driving forces for changes in geographical distribution of Ixodes ricinus ticks in Europe. Parasites and Vectors, 6. https://doi.org/10.1186/1756-3305-6-1 Perez, G., Bournez, L., Boulanger, N., Fite, J., Livoreil, B., McCoy, K., Quillery, E., René-Martellet, M., Bonnet, S. (2023) The distribution, phenology, host range and pathogen prevalence of Ixodes ricinus in France: a systematic map and narrative review. bioRxiv, ver. 1 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2023.04.18.537315 Velez, R., De Meeûs, T., Beati, L., Younsi, H., Zhioua, E., Antunes, S., Domingos, A., Ataíde Sampaio, D., Carpinteiro, D., Moerbeck, L., Estrada-Peña, A., Santos-Silva, M.M., Santos, A.S. (2023) Development and testing of microsatellite loci for the study of population genetics of Ixodes ricinus Linnaeus, 1758 and Ixodes inopinatus Estrada-Peña, Nava & Petney, 2014 (Acari: Ixodidae) in the western Mediterranean region. Acarologia, 63, 356-372. https://doi.org/10.24349/bvem-4h49 | The distribution, phenology, host range and pathogen prevalence of *Ixodes ricinus* in France: a systematic map and narrative review | Grégoire Perez, Laure Bournez, Nathalie Boulanger, Johanna Fite, Barbara Livoreil, Karen D. McCoy, Elsa Quillery, Magalie René-Martellet, and Sarah I. Bonnet | <p style="text-align: justify;">The tick <em>Ixodes ricinus</em> is the most important vector species of infectious diseases in European France. Understanding its distribution, phenology, and host species use, along with the distribution and preva... | | Animal diseases, Behaviour of hosts, infectious agents, or vectors, Disease Ecology/Evolution, Ecohealth, Ecology of hosts, infectious agents, or vectors, Epidemiology, Geography of infectious diseases, Interactions between hosts and infectious ag... | Ana Sofia Santos | 2022-12-06 14:52:44 | ||
14 Feb 2024
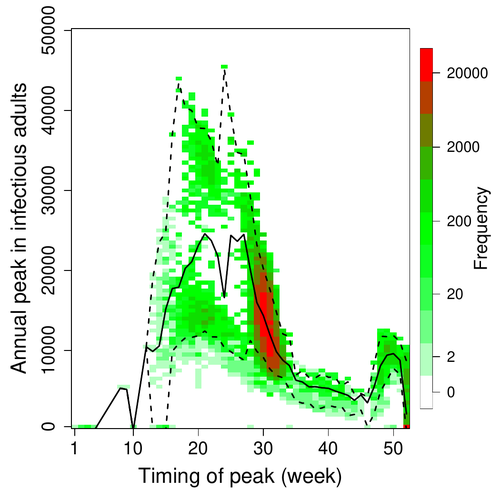
A Bayesian analysis of birth pulse effects on the probability of detecting Ebola virus in fruit batsEpidemiological modeling to optimize the detection of zoonotic viruses in wild (reservoir) speciesRecommended by Aurelien Tellier based on reviews by Hetsron Legrace NYANDJO BAMEN and 1 anonymous reviewer based on reviews by Hetsron Legrace NYANDJO BAMEN and 1 anonymous reviewer
Various species of Ebolavirus have caused, and are still causing, zoonotic outbreaks and public health crises in Africa. Bats have long been hypothesized to be important reservoir populations for a series of viruses such as Hendra or Marburg viruses, the severe acute respiratory syndrome coronavirus (SARS-CoV, SARS-CoV-2) as well as Ebolaviruses [2, 3]. However the ecology of disease dynamics, disease transmission, and coevolution with their natural hosts of these viruses is still poorly understood, despite their importance for predicting novel outbreaks in human or livestock populations. The evidence that bats function as sylvatic reservoirs for Ebola viruses is yet only partial. Indeed, only few serological studies demonstrated the presence of Ebolavirus antibodies in young bats [4], albeit without providing positive controls of viral detection or identifying the viral species (via PCR). There is thus an unexplained discrepancy between serological data and viral detection [2, 4]. In this article, Pleydell et al. [1] use a modeling approach as well as published serological and age-structure (of the bat population) data to calibrate the model simulations. The study starts with the development of an age-structured epidemiological model which includes seasonal birth pulses and waning immunity, both generating pulses of Ebolavirus transmission within a colony of African straw-coloured fruit bats (Eidolon helvum). The epidemiological dynamics of such system of ordinary differential equations can generate annual outbreaks, skipped years or multi-annual cycles up to chaotic dynamics. Therefore, the calibration of the parameters, and the definition of biologically relevant priors, are key. To this aim, the serological data are obtained from a previous study in Cameroon [5], and the age structured of the bat population (birth and mortality) from a population study in Ghana [6]. These data are integrated into the Bayesian analysis and statistical framework to fit the model and generate predictions. In a nutshell, the authors show an overlap between the data and credibility intervals generated by the calibrated model, which thus explains well the seasonality of age-structure, namely changes in pup presence, number of lactating females, or proportion of juveniles in May. The authors can estimate that 76% of adults and 39% of young bats do survive each year, and infections are expected to last one and a half weeks. The epidemiological model predicts that annual birth pulses likely generate annual disease outbreaks, so that weeks 30 to 31 of each year, are predicted to be the best period to isolate the circulating Ebolavirus in this bat population. From the model predictions, the authors estimate the probability to have missed an infectious bat among all the samples tested by PCR being approximately of one per two thousands. The disease dynamics pattern observed in the serology data, and replicated by the model, is likely driven by seasonal pulses of young susceptible bats entering the population. This seasonal birth event increases the viral transmission, resulting in the observed peak of viral prevalence. With the inclusion of immunity waning and antibody persistence, the model results illuminate therefore why previous studies have detected only few positive cases by PCR tests, in contrast to the evidence from serological data. This study provides a first proof of principle that epidemiological modeling, despite its many simplifying assumptions, can be applied to wild species reservoirs of zoonotic diseases in order to optimize the design of field studies to detect viruses. Furthermore, such models can contribute to assess the probability and timing of zoonotic outbreaks in human or livestock populations. This article illustrates one of the manifold applications of mathematical theory of disease epidemiology to optimize sampling of pathogens/parasites or vaccine development and release [7, 8]. The further coupling of such models with population genetics theory and statistical inference methods (using parasite genome data) increasingly provide insights into the adaptation and evolution of parasites to human, crops and livestock populations [9, 10].
References [1] Pleydell D.R.J., Ndong Bass I., Mba Djondzo F.A., Djomsi D.M., Kouanfack C., Peeters M., and J. Cappelle. 2023. A Bayesian analysis of birth pulse effects on the probability of detecting Ebola virus in fruit bats. bioRxiv, ver. 3 peer reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2023.08.10.552777 [2] Caron A., Bourgarel M., Cappelle J., Liégeois F., De Nys H.M., and F. Roger. 2018. Ebola virus maintenance: if not (only) bats, what else? Viruses 10, 549. https://doi.org/10.3390/v10100549 [3] Letko M., Seifert S.N., Olival K.J., Plowright R.K., and V.J. Munster. 2020. Bat-borne virus diversity, spillover and emergence. Nature Reviews Microbiology 18, 461–471. https://doi.org/10.1038/s41579-020-0394-z [4] Leroy E.M., Kumulungui B., Pourrut X., Rouquet P., Hassanin A., Yaba P., Délicat A., Paweska J.T., Gonzalez J.P., and R. Swanepoel. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438, 575–576. https://doi.org/10.1038/438575a [5] Djomsi D.M. et al. 2022. Dynamics of antibodies to Ebolaviruses in an Eidolon helvum bat colony in Cameroon. Viruses 14, 560. https://doi.org/10.3390/v14030560 [6] Peel A.J. et al. 2016. Bat trait, genetic and pathogen data from large-scale investigations of African fruit bats Eidolon helvum. Scientific data 3, 1–11. https://doi.org/10.1038/sdata.2016.49 [7] Nyandjo Bamen H.L., Ntaganda J.M., Tellier A. and O. Menoukeu Pamen. 2023. Impact of imperfect vaccine, vaccine trade-off and population turnover on infectious disease dynamics. Mathematics, 11(5), p.1240. https://doi.org/10.3390/math11051240 [8] Saadi N., Chi Y.L., Ghosh S., Eggo R.M., McCarthy C.V., Quaife M., Dawa J., Jit M. and A. Vassall. 2021. Models of COVID-19 vaccine prioritisation: a systematic literature search and narrative review. BMC medicine, 19, pp.1-11. https://doi.org/10.1186/s12916-021-02190-3 [9] Maerkle, H., John S., Metzger, L., STOP-HCV Consortium, Ansari, M.A., Pedergnana, V. and Tellier, A., 2023. Inference of host-pathogen interaction matrices from genome-wide polymorphism data. bioRxiv, https://doi.org/10.1101/2023.07.06.547816. [10] Gandon S., Day T., Metcalf C.J.E. and B.T. Grenfell. 2016. Forecasting epidemiological and evolutionary dynamics of infectious diseases. Trends in ecology & evolution, 31(10), pp.776-788. https://doi.org/10.1016/j.tree.2016.07.010 | A Bayesian analysis of birth pulse effects on the probability of detecting Ebola virus in fruit bats | David R.J. Pleydell, Innocent Ndong Bass, Flaubert Auguste Mba Djondzo, Dowbiss Meta Djomsi, Charles Kouanfack, Martine Peeters, Julien Cappelle | <p>Since 1976 various species of Ebolavirus have caused a series of zoonotic outbreaks and public health crises in Africa. Bats have long been hypothesised to function as important hosts for ebolavirus maintenance, however the transmission ecology... | | Animal diseases, Disease Ecology/Evolution, Ecohealth, Ecology of hosts, infectious agents, or vectors, Epidemiology, Population dynamics of hosts, infectious agents, or vectors, Reservoirs, Viruses, Zoonoses | Aurelien Tellier | 2023-08-16 16:57:05 | ||
28 Oct 2022

Development of nine microsatellite loci for Trypanosoma lewisi, a potential human pathogen in Western Africa and South-East Asia, and preliminary population genetics analysesPreliminary population genetic analysis of Trypanosoma lewisiRecommended by Annette MacLeod based on reviews by Gabriele Schönian and 1 anonymous reviewerTrypanosoma lewisi is an atypical trypanosome species. Transmitted by fleas, it has a high prevalence and worldwide distribution in small mammals, especially rats [1]. Although not typically thought to infect humans, there has been a number of reports of human infections by T. lewisi in Asia including a case of a fatal infection in an infant [2]. The fact that the parasite is resistant to lysis by normal human serum [3] suggests that many people, especially immunocompromised individuals, may be at risk from zoonotic infections by this pathogen, particularly in regions where there is close contact with T. lewisi-infected rat fleas. Indeed, it is also possible that cryptic T. lewisi infections exist but have hitherto gone undetected. Such asymptomatic infections have been detected for a number of parasitic infections including the related parasite T. b. gambiense [4]. References
[2] Truc P, Büscher P, Cuny G, Gonzatti MI, Jannin J, Joshi P, Juyal P, Lun Z-R, Mattioli R, Pays E, Simarro PP, Teixeira MMG, Touratier L, Vincendeau P, Desquesnes M (2013) Atypical Human Infections by Animal Trypanosomes. PLOS Neglected Tropical Diseases, 7, e2256. https://doi.org/10.1371/journal.pntd.0002256 [3] Lun Z-R, Wen Y-Z, Uzureau P, Lecordier L, Lai D-H, Lan Y-G, Desquesnes M, Geng G-Q, Yang T-B, Zhou W-L, Jannin JG, Simarro PP, Truc P, Vincendeau P, Pays E (2015) Resistance to normal human serum reveals Trypanosoma lewisi as an underestimated human pathogen. Molecular and Biochemical Parasitology, 199, 58–61. https://doi.org/10.1016/j.molbiopara.2015.03.007 [4] Büscher P, Bart J-M, Boelaert M, Bucheton B, Cecchi G, Chitnis N, Courtin D, Figueiredo LM, Franco J-R, Grébaut P, Hasker E, Ilboudo H, Jamonneau V, Koffi M, Lejon V, MacLeod A, Masumu J, Matovu E, Mattioli R, Noyes H, Picado A, Rock KS, Rotureau B, Simo G, Thévenon S, Trindade S, Truc P, Reet NV (2018) Do Cryptic Reservoirs Threaten Gambiense-Sleeping Sickness Elimination? Trends in Parasitology, 34, 197–207. https://doi.org/10.1016/j.pt.2017.11.008 [5] Ségard A, Roméro A, Ravel S, Truc P, Gauthier D, Gauthier P, Dossou H-J, Sylvestre B, Houéménou G, Morand S, Chaisiri K, Noûs C, De Meeûs T (2022) Development of nine microsatellite loci for Trypanosoma lewisi, a potential human pathogen in Western Africa and South-East Asia, and preliminary population genetics analyses. Zenodo, 6460010, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.6460010 | Development of nine microsatellite loci for Trypanosoma lewisi, a potential human pathogen in Western Africa and South-East Asia, and preliminary population genetics analyses | Adeline Ségard, Audrey Romero, Sophie Ravel, Philippe Truc, Gauthier Dobigny, Philippe Gauthier, Jonas Etougbetche, Henri-Joel Dossou, Sylvestre Badou, Gualbert Houéménou, Serge Morand, Kittipong Chaisiri, Camille Noûs, Thierry deMeeûs | <p><em>Trypanosoma lewisi</em> belongs to the so-called atypical trypanosomes that occasionally affect humans. It shares the same hosts and flea vector of other medically relevant pathogenic agents as Yersinia pestis, the agent of plague. Increasi... | | Animal diseases, Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Eukaryotic pathogens/symbionts, Evolution of hosts, infectious agents, or vectors, Microbiology of infections, Parasites, Population genetics of hosts, in... | Annette MacLeod | 2022-04-21 17:04:37 | ||
17 Jan 2024
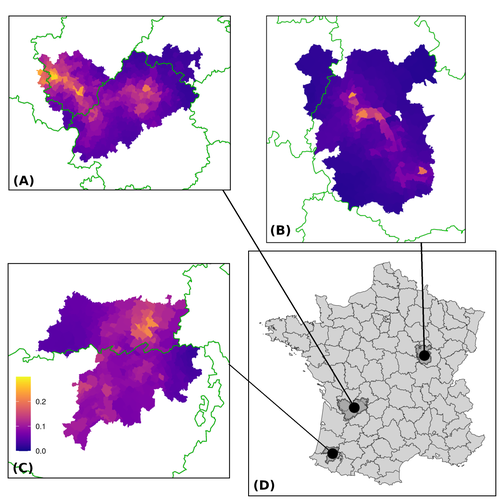
Assessing the dynamics of Mycobacterium bovis infection in three French badger populationsFrom disease surveillance to public action. Re-inforcing both epidemiological surveillance and data analysis: an illustration with Mycobacterium bovisRecommended by Jean-Francois Guégan based on reviews by Rowland Kao and 1 anonymous reviewerMycobacterium bovis, also called M. tuberculosis var. bovis, is a bacterium belonging to the M. tuberculosis complex (i.e., MTBC) and which can cause through zoonotic transmission another form of human tuberculosis (Tb). It is above all the agent of bovine tuberculosis (i.e., bTb) which affects not only cattle (wild or farmed) but also a large diversity of other wild mammals worldwide. An increasing number of infected animal cases are being discovered in many regions of the world, thus raising the problem of tuberculosis transmission, including to humans, more complex than previously thought. Efforts have been made in terms of vaccination or culling of populations of host carrier species, such as the badger for example, however leading to consequences of greater dispersion of the infectious agent. M. bovis shows a more or less significant capacity to persist outside its hosts, particularly in the environment under certain abiotic and biotic conditions. This bacillus can be transmitted and spread in many ways, including through aerosol, mucus and sputum, urine and feces, by direct contact with infected animals, their dead bodies or rather via their excreta or by inhalation of aerosols, depending on the host species concerned. In this paper, Calenge and his collaborators (Callenge et al. 2024) benefited from a national surveillance program on M. bovis cases in wild species, set up in 2011 in France, i.e., Sylvatub, for detecting and monitoring M. bovis infection in European badger (Meles meles) populations. Sylvatub is a participatory program involving both national and local stakeholder systems in order to determine changes in bTb infection levels in domestic and wild animal species. This original work had two aims: to describe spatial disease dynamics in the three clusters under scrutiny using a complex Bayesian model; and to develop indicators for the monitoring of the M. bovis infection by stakeholders and decision-makers of the program. This paper is timely and very comprehensive. In this cogent study, the authors illustrate this point by using epidemiological surveillance to obtain large amounts of data (which is generally lacking in human epidemiology, but more dramatically lacking in animal epidemiology) and a highly sophisticated biostatistical analysis (Callenge et al. 2024). It is in itself a demonstration of the current capabilities of population dynamics applied to infectious disease situations, in this case animal, in the rapidly developing discipline of disease ecology and evolution. One of the aims of the study is to propose statistical models that can be used by the different stakeholders in charge, for instance, of wildlife conservation or the regional or State veterinary services to assess disease risk in the most affected regions. References Assel AKHMETOVA, Jimena GUERRERO, Paul McADAM, Liliana CM SALVADOR, Joseph CRISPELL, John LAVERY, Eleanor PRESHO, Rowland R KAO, Roman BIEK, Fraser MENZIES, Nigel TRIMBLE, Roland HARWOOD, P Theo PEPLER, Katarina ORAVCOVA, Jordon GRAHAM, Robin SKUCE, Louis DU PLESSIS, Suzan THOMPSON, Lorraine WRIGHT, Andrew W BYRNE, Adrian R ALLEN. 2023. Genomic epidemiology of Mycobacterium bovis infection in sympatric badger and cattle populations in Northern Ireland. Microbial Genomics 9: mgen001023. https://doi.org/10.1099/mgen.0.001023 Roman BIEK, Anthony O’HARE, David WRIGHT, Tom MALLON, Carl McCORMICK, Richard J ORTON, Stanley McDOWELL, Hannah TREWBY, Robin A SKUCE, Rowland R KAO. 2012. Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathogens 8: e1003008. https://doi.org/10.1371/journal.ppat.1003008 Clément CALENGE, Ariane PAYNE, Edouard REVEILLAUD, Céline RICHOMME, Sébastien GIRARD, Stephanie DESVAUX. 2024. Assessing the dynamics of Mycobacterium bovis infection in three French badger populations. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2023.05.31.543041 Marc CHOISY, Pejman ROHANI. 2006. Harvesting can increase severity of wildlife disease epidemics. Proceedings of the Royal Society, London, Ser. B 273: 2025-2034. https://doi.org/10.1098/rspb.2006.3554 Shannon C DUFFY, Sreenidhi SRINIVASAN, Megan A SCHILLING, Tod STUBER, Sarah N DANCHUK, Joy S MICHAEL, Manigandan VENKATESAN, Nitish BANSAL, Sushila MAAN, Naresh JINDAL, Deepika CHAUDHARY, Premanshu DANDAPAT, Robab KATANI, Shubhada CHOTHE, Maroudam VEERASAMI, Suelee ROBBE-AUSTERMAN, Nicholas JULEFF, Vivek KAPUR, Marcel A BEHR. 2020. Reconsidering Mycobacterium bovis as a proxy for zoonotic tuberculosis: a molecular epidemiological surveillance study. Lancet Microbe 1: e66-e73. https://doi.org/10.1016/S2666-5247(20)30038-0 Jean-François GUEGAN. 2019. The nature of ecology of infectious disease. The Lancet Infectious Diseases 19. https://doi.org/10.1016/s1473-3099(19)30529-8 Brandon H HAYES, Timothée VERGNE, Mathieu ANDRAUD, Nicolas ROSE. 2023. Mathematical modeling at the livestock-wildlife interface: scoping review of drivers of disease transmission between species. Frontiers in Veterinary Science 10: 1225446. https://doi.org/10.3389/fvets.2023.1225446 David KING, Tim ROPER, Douglas YOUNG, Mark EJ WOOLHOUSE, Dan COLLINS, Paul WOOD. 2007. Bovine tuberculosis in cattle and badgers. Report to Secretary of State about tuberculosis in cattle and badgers. London, UK. https://www.bovinetb.info/docs/RBCT_david_%20king_report.pdf Robert MM SMITH , Francis DROBNIEWSKI, Andrea GIBSON, John DE MONTAGUE, Margaret N LOGAN, David HUNT, Glyn HEWINSON, Roland L SALMON, Brian O’NEILL. 2004. Mycobacterium bovis Infection, United Kingdom. Emerging Infectious Diseases 10: 539-541. https://doi.org/10.3201/eid1003.020819 | Assessing the dynamics of *Mycobacterium bovis* infection in three French badger populations | Clement CALENGE, Ariane PAYNE, Edouard REVEILLAUD, Celine RICHOMME, Sebastien GIRARD, Stephanie DESVAUX | <p>The Sylvatub system is a national surveillance program established in 2011 in France to monitor infections caused by <em>Mycobacterium bovis</em>, the main etiologic agent of bovine tuberculosis, in wild species. This participatory program, inv... | | Animal diseases, Ecohealth, Ecology of hosts, infectious agents, or vectors, Epidemiology, Geography of infectious diseases, Pathogenic/Symbiotic Bacteria, Zoonoses | Jean-Francois Guégan | 2023-06-05 10:50:49 | ||
03 Nov 2023

Longitudinal Survey of Astrovirus infection in different bat species in Zimbabwe: Evidence of high genetic Astrovirus diversityHigh diversity and evidence for inter-species transmission in astroviruses surveyed from bats in ZibabwaeRecommended by Tim James based on reviews by 2 anonymous reviewersMost infectious diseases of humans are zoonoses, and many of these come from particularly species diverse reservoir taxa, such as bats, birds, and rodents (1). Because of our changing landscape, there is increased exposure of humans to wildlife diseases reservoirs, yet we have little basic information about prevalence, hotspots, and transmission factors of most zoonotic pathogens. Viruses are particularly worrisome as a public health risk due to their fast mutation rates and well-known cross-species transmission abilities. There is a global push to better survey wildlife for viruses (2), but these studies are difficult, and the problem is vast. Astroviruses (AstVs) comprise a diverse family of ssRNA viruses known from mammals and birds. Astroviruses can cause gastroenteritis in humans and are more common in elderly and young children, but the relationship of human to non-human Astroviridae as well as transmission routes are unclear. AstVs have been detected at high prevalence in bats in multiple studies (3,4), but it is unclear what factors, such as co-infecting viruses and bat reproductive phenology, influence viral shedding and prevalence. References 1. Mollentze N, Streicker DG. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proceedings of the National Academy of Sciences. 2020 Apr 28;117(17):9423-30. https://doi.org/10.1073/pnas.1919176117 | Longitudinal Survey of Astrovirus infection in different bat species in Zimbabwe: Evidence of high genetic Astrovirus diversity | Vimbiso Chidoti, Helene De Nys, Malika Abdi, Getrudre Mashura, Valerie Pinarello, Ngoni Chiweshe, Gift Matope, Laure Guerrini, Davies Pfulenyi, Julien Cappelle, Ellen Mwandiringana, Dorothee Misse, Gori Elizabeth, Mathieu Bourgarel, Florian Liegeois | <p>Astroviruses (AstVs) have been discovered in over 80 animal species including diverse bat species and avian species. A study on Astrovirus circulation and diversity in different insectivorous and frugivorous chiropteran species roosting in tree... | | Animal diseases, Epidemiology, Molecular genetics of hosts, infectious agents, or vectors, Reservoirs, Viruses, Zoonoses | Tim James | 2023-04-18 14:58:43 | ||
21 Jul 2022

Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV)Understanding the in vitro evolution of Cyprinid herpesvirus 3 (CyHV-3), a story of structural variations that can lead to the design of attenuated virus vaccinesRecommended by Jorge Amich based on reviews by Lucie Cappuccio and Veronique Hourdel
Structural variations (SVs) play a key role in viral evolution, and therefore they are also important for infection dynamics. However, the contribution of structural variations to the evolution of double-stranded viruses is limited. This knowledge can help to understand the population dynamics and might be crucial for the future development of viral attenuated vaccines. In this study, Fuandila et al (1) use the Cyprinid herpesvirus 3 (CyHV-3), commonly known as koi herpesvirus (KHV), to investigate the variability and contribution of structural variations (SV) for viral evolution after 99 passages in vitro. This virus, with the largest genome among herperviruses, causes a lethal infection in common carp and koi associated with mortalities up to 95% (2). Interestingly, KHV infections are caused by haplotype mixtures, which possibly are a source of genome diversification, but make genomic comparisons more difficult. The authors have used ultra-deep long-read sequencing of two passages, P78 and P99, which were previously described to have differences in virulence. They have found a surprisingly high and wide distribution of SVs along the genome, which were enriched in inversion and deletion events and that often led to defective viral genomes. Although it is known that these defective viral genomes negatively impact viral replication, their implications for virus persistence are still unclear. Subsequently, the authors concentrated on the virulence-relevant region ORF150, which was found to be different in P78 (deletion in 100% of the reads) and P99 (reference-like haplotype). To understand this loss and gain of full ORF150, they searched for SV turn-over in 10 intermediate passages. This analysis revealed that by passage 10 deleted and inverted (attenuated) haplotypes had already appeared, steadily increased frequency until P78, and then completely disappeared between P78 and P99. This is a striking result that raises new questions as to how this clearance occurs, which is really important as these reversions may result in undesirable increases in virulence of live-attenuated vaccines. We recommend this preprint because its use of ultra-deep long-read sequencing has permitted to better understand the role of SV diversity and dynamics in viral evolution. This study shows an unexpectedly high number of structural variations, revealing a novel source of virus diversification and confirming the different mixtures of haplotypes in different passages, including the gain of function. This research provides basic knowledge for the future design of live-attenuated vaccines, to prevent the reversion to virulent viruses. References (1) Fuandila NN, Gosselin-Grenet A-S, Tilak M-K, Bergmann SM, Escoubas J-M, Klafack S, Lusiastuti AM, Yuhana M, Fiston-Lavier A-S, Avarre J-C, Cherif E (2022) Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV). bioRxiv, 2022.03.10.483410, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.03.10.483410 (2) Sunarto A, McColl KA, Crane MStJ, Sumiati T, Hyatt AD, Barnes AC, Walker PJ. Isolation and characterization of koi herpesvirus (KHV) from Indonesia: identification of a new genetic lineage. Journal of Fish Diseases, 34, 87-101. https://doi.org/10.1111/j.1365-2761.2010.01216.x | Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV) | Nurul Novelia Fuandila, Anne-Sophie Gosselin-Grenet, Marie-Ka Tilak, Sven M Bergmann, Jean-Michel Escoubas, Sandro Klafack, Angela Mariana Lusiastuti, Munti Yuhana, Anna-Sophie Fiston-Lavier, Jean-Christophe Avarre, Emira Cherif | <p style="text-align: justify;">Structural variations (SVs) constitute a significant source of genetic variability in virus genomes. Yet knowledge about SV variability and contribution to the evolutionary process in large double-stranded (ds)DNA v... | | Animal diseases, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Viruses | Jorge Amich | Lucie Cappuccio, Veronique Hourdel | 2022-03-11 10:50:50 | |
23 Jan 2023

Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolenseWhole genome transcriptome reveals metabolic and immune susceptibility factors for Trypanosoma congolense infection in West-African livestockRecommended by Concepción Marañón based on reviews by 2 anonymous reviewersAfrican trypanosomiasis is caused by to the infection of a protozoan parasite of the Trypanosoma genus. It is transmitted by the tsetse fly, and is largely affecting cattle in the sub-humid areas of Africa, causing a high economic impact. However, not all the bovine strains are equally susceptible to the infection (1). In order to dissect the mechanisms underlying susceptibility to African trypanosoma infection, Peylhard et al (2) performed blood transcriptional profiles of trypanotolerant, trypanosensitive and mixed cattle breeds, before and after experimental infection with T. congolense. First of all, the authors have characterized the basal transcriptional profiles in the blood of the different breeds under study, which could be classified in a wide array of functional pathways. Of note, after infection some pathways were consistently enriched in all the group tested. Among them, the immune system-related ones were again on the top functions reported. The search for specific canonical pathways pointed to a prominent role of lipid and cholesterol-related pathways, as well as mitochondrial function and B and T lymphocyte activation. However, the analysis of infected animals demonstrated that trypanosusceptible animals showed a stronger transcriptomic reprogramming, highly enriched in specific metabolic and immunological pathways. It is worthy to highlight striking differences in genes involved in immune signal transduction, cytokines and markers of different leukocyte subpopulations. This work represents undoubtedly a significant momentum in the field, since the authors explore in deep a wide panel of cattle breeds representing the majority of West-African taurine and zebu in a systematic way. Since the animals were studied at different timepoints after infection, future longitudinal analyses of these datasets will be providing a precious insight on the kinetics of immune and metabolic reprogramming associated with susceptibility and tolerance to African trypanosoma infection, widening the application of this interesting study into new therapeutic interventions. References 1. Berthier D, Peylhard M, Dayo G-K, Flori L, Sylla S, Bolly S, Sakande H, Chantal I, Thevenon S (2015) A Comparison of Phenotypic Traits Related to Trypanotolerance in Five West African Cattle Breeds Highlights the Value of Shorthorn Taurine Breeds. PLOS ONE, 10, e0126498. https://doi.org/10.1371/journal.pone.0126498 2. Peylhard M, Berthier D, Dayo G-K, Chantal I, Sylla S, Nidelet S, Dubois E, Martin G, Sempéré G, Flori L, Thévenon S (2022) Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense. bioRxiv, 2022.06.10.495622, ver. 2 peer-reviewed and recommended by Peer Community Infections. https://doi.org/10.1101/2022.06.10.495622. | Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense | Moana Peylhard, David Berthier, Guiguigbaza-Kossigan Dayo, Isabelle Chantal, Souleymane Sylla, Sabine Nidelet, Emeric Dubois, Guillaume Martin, Guilhem Sempéré, Laurence Flori, Sophie Thévenon | <p>Animal African trypanosomosis, caused by blood protozoan parasites transmitted mainly by tsetse flies, represents a major constraint for millions of cattle in sub-Saharan Africa. Exposed cattle include trypanosusceptible indicine breeds, severe... | | Animal diseases, Genomics, functional genomics of hosts, infectious agents, or vectors, Resistance/Virulence/Tolerance | Concepción Marañón | Anonymous, Anonymous | 2022-06-14 17:06:57 | |
14 Dec 2022

Transcriptome responses of the aphid vector Myzus persicae are shaped by identities of the host plant and the virusHow do multiple host plants and virus species challenge aphid molecular machinery?Recommended by Sebastien Massart based on reviews by Juan José Lopez Moya and Michelle HeckThe impact of virus infection of a plant on an aphid’s behaviour has been observed in many studies [1]. Indeed, virus infection can alter plant biochemistry through the emission of volatile organic compounds and plant tissue content modification. These alterations can further impact the interactions between plants and aphids. However, although it is a well-known phenomenon, very few studies have explored the consequences of plant virus infection on the gene expression of aphids to understand better the aphid’s manipulation by the plant virus. In this context, the recommended study [2] reports a comprehensive transcriptomic analysis of the genes expressed by one aphid species, Myzus persicae, a vector of several plant viruses, when feeding on plants. Michelle Heck underlined how significant this study is for comprehending the molecular bases of aphid-vector manipulation by plant viruses (see below). Interestingly, the study design has integrated several factors that might influence the gene expression of M. persicae when feeding on the plant. Indeed, the authors investigated the effect of two plant species (Arabidopsis thaliana and Camelia sativa) and two virus species [turnip yellows virus (TuYV) and cauliflower mosaic virus (CaMV)]. Noteworthy, the transmission mode of TuYV is circulative and persistent, while CaMV is transmitted by a semi-persistent non-circulative mode. As Juan José Lopez Moya mentioned, multiple comparisons allowed the identification of the different responses of aphids in front of different host plants infected or not by different viruses (see below). This publication is complementary to a previous publication from the same team focusing on plant transcriptome analysis [3]. Thanks to their experimental design, the authors identified genes commonly deregulated by both viruses and/or both plant species and deregulated genes by a single virus or a single plant. Figure 4 nicely summarizes the number of deregulated genes. A thorough discussion on the putative role of deregulated genes in different conditions gave a comprehensive follow-up of the results and their impact on the current knowledge of plant-virus-vector interactions. This study has now opened the gate to promising research focusing on the functional validation of the identified genes while also narrowing the study from the body to the tissue level. References: 1. Carr JP, Tungadi T, Donnelly R, Bravo-Cazar A, Rhee S-J, Watt LG, Mutuku JM, Wamonje FO, Murphy AM, Arinaitwe W, Pate AE, Cunniffe NJ, Gilligan CA (2020) Modelling and manipulation of aphid-mediated spread of non-persistently transmitted viruses. Virus Research, 277, 197845. https://doi.org/10.1016/j.virusres.2019.197845 2. Chesnais Q, Golyaev V, Velt A, Rustenholz C, Verdier M, Brault V, Pooggin MM, Drucker M (2022) Transcriptome responses of the aphid vector Myzus persicae are shaped by identities of the host plant and the virus. bioRxiv , 2022.07.18.500449, ver. 5 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.07.18.500449 3. Chesnais Q, Golyaev V, Velt A, Rustenholz C, Brault V, Pooggin MM, Drucker M (2022) Comparative Plant Transcriptome Profiling of Arabidopsis thaliana Col-0 and Camelina sativa var. Celine Infested with Myzus persicae Aphids Acquiring Circulative and Noncirculative Viruses Reveals Virus- and Plant-Specific Alterations Relevant to Aphid Feeding Behavior and Transmission. Microbiology Spectrum, 10, e00136-22. https://doi.org/10.1128/spectrum.00136-22 | Transcriptome responses of the aphid vector *Myzus persicae* are shaped by identities of the host plant and the virus | Quentin Chesnais, Victor Golyaev, Amandine Velt, Camille Rustenholz, Maxime Verdier, Véronique Brault, Mikhail M. Pooggin, Martin Drucker | <p style="text-align: justify;"><strong>Background:</strong> Numerous studies have documented modifications in vector orientation behavior, settling and feeding behavior, and/or fecundity and survival due to virus infection in host plants. These a... | | Behaviour of hosts, infectious agents, or vectors, Cell biology of hosts, infectious agents, or vectors, Molecular biology of infections, Physiology of hosts, infectious agents, or vectors, Phytopathology, Plant diseases, Vectors, Viruses | Sebastien Massart | 2022-07-19 15:24:14 | ||
24 Jan 2024
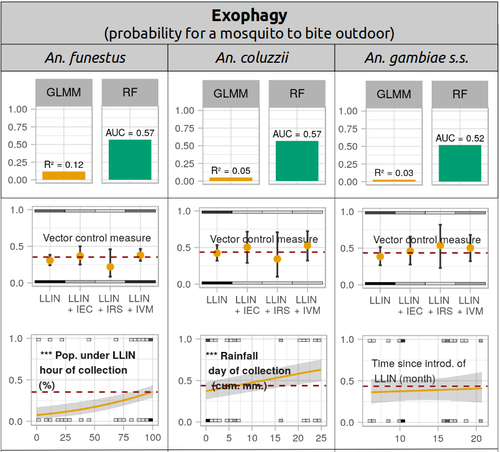
Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictabilityLarge and complete datasets, and modelling reveal the major determinants of physiological and behavioral insecticide resistance of malaria vectorsRecommended by Thierry DE MEEÛS based on reviews by Haoues Alout and 1 anonymous reviewer
Parasites represent the most diverse and adaptable ecological group of the biosphere (Timm & Clauson, 1988; De Meeûs et al., 1998; Poulin & Morand, 2000; De Meeûs & Renaud, 2002). The human species is known to considerably alter biodiversity, though it hosts, and thus sustains the maintenance of a spectacular diversity of parasites (179 species for eukaryotic species only) (De Meeûs et al., 2009). Among these, the five species of malaria agents (genus Plasmodium) remain a major public health issue around the world. Plasmodium falciparum is the most prevalent and lethal of these (Liu et al., 2010). With a pick of up to 2 million deaths due to malaria in 2004, deaths decreased to around 1 million in 2010 (Murray et al., 2012), to reach 619,000 in 2021, most of which in sub-Saharan Africa, and 79% of which were among children aged under 5 years (World Health Organization, 2022). As stressed by Taconet et al. (2023), reduction in malaria deaths is attributable to control measures, in particular against its vectors (mosquitoes of the genus Anopheles). Nevertheless, the success of vector control is hampered by several factors (biological, environmental and socio-economic), and in particular by the great propensity of targeted mosquitoes to evolve physiological or behavioral avoidance of anti-vectorial measures. In their paper Taconet et al. (2023) aims at understanding what are the main factors that determine the evolution of insecticide resistance in several malaria vectors, in relation to the biological determinisms of behavioral resistance and how fast such evolutions take place. To tackle these objectives, authors collected an impressive amount of data in two rural areas of West Africa. With appropriate modeling, Taconet et al. discovered, among many other results, a predominant role of public health measures, as compared to agricultural practices, in the evolution of physiological resistance. They also found that mosquito foraging activities are mostly explained by host availability and climate, with a poor, if any, association with genetic markers of physiological resistance to insecticides. These findings represent an important contribution to the field and should help at designing more efficient control strategies against malaria.
References De Meeûs T, Michalakis Y, Renaud F (1998) Santa Rosalia revisited: or why are there so many kinds of parasites in “the garden of earthly delights”? Parasitology Today, 14, 10–13. https://doi.org/10.1016/S0169-4758(97)01163-0 De Meeûs T, Prugnolle F, Agnew P (2009) Asexual reproduction in infectious diseases. In: Lost Sex: The Evolutionary Biology of Parthenogenesis (eds Schön I, Martens K, van Dijk P), pp. 517-533. Springer, NY. https://doi.org/10.1007/978-90-481-2770-2_24 De Meeûs T, Renaud F (2002) Parasites within the new phylogeny of eukaryotes. Trends in Parasitology, 18, 247–251. https://doi.org/10.1016/S1471-4922(02)02269-9 Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF, Ndjango JB, Sanz CM, Morgan DB, Locatelli S, Gonder MK, Kranzusch PJ, Walsh PD, Delaporte E, Mpoudi-Ngole E, Georgiev AV, Muller MN, Shaw GM, Peeters M, Sharp PM, Rayner JC, Hahn BH (2010) Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature, 467, 420–425. https://doi.org/10.1038/nature09442 Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. The Lancet, 379, 413–431. https://doi.org/10.1016/S0140-6736(12)60034-8 Poulin R, Morand S (2000) The diversity of parasites. Quarterly Review of Biology, 75, 277–293. https://doi.org/10.1086/393500 Taconet P, Soma DD, Zogo B, Mouline K, Simard F, Koffi AA, Dabire RK, Pennetier C, Moiroux N (2023) Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictability. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.20.504631 Timm RM, Clauson BL (1988) Coevolution: Mammalia. In: 1988 McGraw-Hill yearbook of science & technology, pp. 212–214. McGraw-Hill Book Company, New York. World Health Organization (2022) World malaria report 2022. Geneva: World Health Organization; 2022. Licence: CC BY-NC-SA 3.0 IGO. https://iris.who.int/bitstream/handle/10665/365169/9789240064898-eng.pdf?sequence=1.
| Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictability | Paul Taconet, Dieudonné Diloma Soma, Barnabas Zogo, Karine Mouline, Frédéric Simard, Alphonsine Amanan Koffi, Roch Kounbobr Dabiré, Cédric Pennetier, Nicolas Moiroux | <p>Insecticide resistance and behavioural adaptation of malaria mosquitoes affect the efficacy of long-lasting insecticide nets - currently the main tool for malaria vector control. To develop and deploy complementary, efficient and cost-effective... | | Behaviour of hosts, infectious agents, or vectors, Ecology of hosts, infectious agents, or vectors, Pesticide resistance, Population genetics of hosts, infectious agents, or vectors, Vectors | Thierry DE MEEÛS | Haoues Alout, Anonymous | 2023-07-03 11:29:10 | |
06 Apr 2023

Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populationsChanges in aggressiveness in pathotypes of wheat leaf rustRecommended by Pierre Gladieux based on reviews by 2 anonymous reviewersUnderstanding the ecological and evolutionary factors underlying the spread of new fungal pathogen populations can inform the development of more effective management strategies. In plant pathology, pathogenicity is generally presented as having two components: ‘virulence’ (qualitative pathogenicity) and aggressiveness (quantitative pathogenicity). Changes in virulence in response to the deployment of new resistant varieties are a major driver of the spread of new populations (called pathotypes, or races) in modern agrosystems, and the genomic (i.e. proximal) and eco-evolutionary (i.e. ultimate) factors underlying these changes are well-documented [1,2,3]. By contrast, the role of changes in aggressiveness in the spread of pathotypes remains little known [4]. The study by Cécilia Fontyn and collaborators [5] set out to characterize changes in aggressiveness for isolates of two pathotypes of the wheat leaf rust (Puccinia triticina) that have been dominant in France during the 2005-2016 period. Isolates were genetically characterized using multilocus microsatellite typing and phenotypically characterized for three components of aggressiveness on wheat varieties: infection efficiency, latency period, and sporulation capacity. Using experiments that represent quite a remarkable amount of work and effort, Fontyn et al. showed that each dominant pathotype consisted of several genotypes, including common genotypes whose frequency changed over time. For each pathotype, the genotypes that were more common initially were replaced by a more aggressive genotype. Together, these results show that changes in the genetic composition of populations of fungal plant pathogens can be associated with, and may be caused by, changes in the quantitative components of pathogenicity. This study also illustrates how extensive, decade-long monitoring of fungal pathogen populations, such as the one conducted for wheat leaf rust in France, represents a very valuable resource for research. REFERENCES [1] Brown, J. K. (1994). Chance and selection in the evolution of barley mildew. Trends in Microbiology, 2(12), 470-475. https://doi.org/10.1016/0966-842x(94)90650-5 [2] Daverdin, G., Rouxel, T., Gout, L., Aubertot, J. N., Fudal, I., Meyer, M., Parlange, F., Carpezat, J., & Balesdent, M. H. (2012). Genome structure and reproductive behaviour influence the evolutionary potential of a fungal phytopathogen. PLoS Pathogens, 8(11), e1003020. https://doi.org/10.1371/journal.ppat.1003020 [3] Gladieux, P., Feurtey, A., Hood, M. E., Snirc, A., Clavel, J., Dutech, C., Roy, M., & Giraud, T. (2015). The population biology of fungal invasions.Molecular Ecology, 24(9), 1969-86. https://doi.org/10.1111/mec.13028 [4] Fontyn, C., Zippert, A. C., Delestre, G., Marcel, T. C., Suffert, F., & Goyeau, H. (2022). Is virulence phenotype evolution driven exclusively by Lr gene deployment in French Puccinia triticina populations?. Plant Pathology, 71(7), 1511-1524. https://doi.org/10.1111/ppa.13599 [5] Fontyn, C., Meyer, K. J., Boixel, A. L., Delestre, G., Piaget, E., Picard, C., Suffer, F., Marcel, T.C., & Goyeau, H. (2022). Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations. bioRxiv, 2022.08.29.505401, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.29.505401 | Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations | Cécilia FONTYN, Kevin JG MEYER, Anne-Lise BOIXEL, Ghislain DELESTRE, Emma PIAGET, Corentin PICARD, Frédéric SUFFERT, Thierry C MARCEL, Henriette GOYEAU | <p style="text-align: justify;">Plant pathogens are constantly evolving and adapting to their environment, including their host. Virulence alleles emerge, and then increase, and sometimes decrease in frequency within pathogen populations in respon... | | Coevolution, Epidemiology, Evolution of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Pathogenic/Symbiotic Fungi, Phytopathology, Plant diseases, Population dynamics of hosts, infectious agents, or... | Pierre Gladieux | Emerson Del Ponte , Jacqui Shykoff, Leïla Bagny Beilhe , Alexey Mikaberidze | 2022-09-29 20:01:57 |




