
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender▲ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
08 Dec 2022
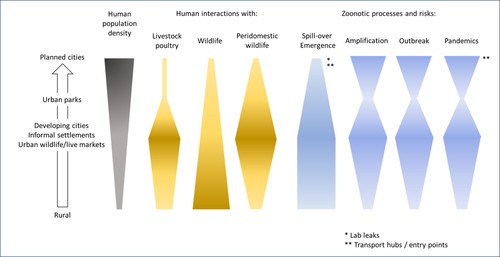
Zoonotic emergence at the animal-environment-human interface: the forgotten urban socio-ecosystemsZoonotic emergence and the overlooked case of citiesRecommended by Etienne Waleckx based on reviews by Eric Dumonteil, Nicole L. Gottdenker and 1 anonymous reviewerZoonotic pathogens, those transmitted from animals to humans, constitute a major public health risk with high associated global economic costs. Diseases associated with these pathogens represent more than 60% of emerging infectious diseases and predominantly originate in wildlife (1). Over the last decades, the emergence and re-emergence of zoonotic pathogens have led to an increasing number of epidemics, as illustrated by the current Covid-19 pandemic. There is ample evidence that human impact on native ecosystems such as deforestation, agricultural development, and urbanization, is linked to spillover of pathogens from animals to humans (2). However, research and calls to action have mainly focused on the importance of surveillance and prevention of zoonotic emergences along landscape interfaces, with special emphasis on tropical forests and agroecosystems, and studies and reviews pointing out the zoonotic risk associated with cities are scarce. Additionally, cities are sometimes wrongly seen as one homogeneous ecosystem, almost exclusively human, with a Northern hemisphere-biased perception of what a city is, which fails to take into account the ecological and socio-economic diversities that can constitute an urban area. Here, Dobigny and Morand (3) aim to draw attention to the importance of urban ecosystems in zoonotic risk and advocate that further attention should be paid to urban, peri-urban and suburban areas. In this well-organized and well-documented review, the authors show, using updated literature, that cities are places where massive contacts occur between wildlife, domestic animals, and human inhabitants (thus constituting spillover opportunities), and that it is even likely that human and wildlife contact in urban centers is more prevalent than in wild areas, perhaps contrary to intuition. Indeed, cities currently constitute the most important environment of human life and are places for millions of close interactions between humans and animals, including pets and domestic animals, wild animals through the intrusion of wild urban-adapted species (e.g., some bat, rodent, or bird species among others), manipulation and consumption of wildlife meat, and the existence of wildlife meat markets, which all constitute a major risk for zoonotic spillover. In cities, lab escapees of zoonotic pathogens also exist, and trends of adaptation to urban ecological conditions of many vectors of primary health importance is also a concern. The authors further argue that cities are predominant places for both epidemic amplification of human-human transmitted pathogens, because they are places with high human densities and population growth, and for dissemination of reservoirs, vectors and pathogens, as they are transport hubs. Dobigny & Morand further predict, likely correctly, that cities may be important places for pathogen evolution. Finally, they propose actions and recommendations to limit the risk of zoonotic spillover events from urban ecosystems and future directions for research aiming at assessing this risk. The reviewers found the manuscript well-organized and presented, timely, and bringing novel contributions to the field of zoonotic emergence. I strongly recommend this article, which should benefit a large audience, particularly in the context of the current Covid-19 pandemics and the ongoing One Health initiatives aiming at preventing future zoonotic disease emergence (4). References (1) Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P (2008) Global trends in emerging infectious diseases. Nature, 451, 990–993. https://doi.org/10.1038/nature06536 (2) White RJ, Razgour O (2020) Emerging zoonotic diseases originating in mammals: a systematic review of effects of anthropogenic land-use change. Mammal Review, 50, 336–352. https://doi.org/10.1111/mam.12201 (3) Dobigny G, Morand S (2022) Zoonotic emergence at the animal-environment-human interface: the forgotten urban socio-ecosystems. Zenodo, 6444776, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.6444776 (4) Morand S, Lajaunie C (2021) Biodiversity and COVID-19: A report and a long road ahead to avoid another pandemic. One Earth, 4, 920–923. https://doi.org/10.1016/j.oneear.2021.06.007 | Zoonotic emergence at the animal-environment-human interface: the forgotten urban socio-ecosystems | Dobigny, G. & Morand, S. | <p style="text-align: justify;">Zoonotic emergence requires spillover from animals to humans, hence animal-human interactions. A lot has been emphasized on human intrusion into wild habitats (e.g., deforestation, hunting) and the development of ag... | | Disease Ecology/Evolution, Ecohealth, Ecology of hosts, infectious agents, or vectors, Evolution of hosts, infectious agents, or vectors, One Health, Zoonoses | Etienne Waleckx | 2022-04-11 11:39:11 | ||
19 Feb 2024

Population genetics of Glossina palpalis gambiensis in the sleeping sickness focus of Boffa (Guinea) before and after eight years of vector control: no effect of control despite a significant decrease of human exposure to the diseaseReaching the last miles for transmission interruption of sleeping sickness in Guinea: follow-up of achievements and policy making using microsatellites-based population geneticsRecommended by Hugues Nana Djeunga based on reviews by Fabien HALKETT and 2 anonymous reviewers based on reviews by Fabien HALKETT and 2 anonymous reviewers
Thanks to the coordinated and sustained efforts of national control programs, the World Health Organization (WHO), bilateral cooperation and nongovernmental organizations, the incidence of Human African Trypanosomiasis (HAT), better known as sleeping sickness, has drastically decreased during the last two decades (WHO, 2023a). Indeed, between 1999 and 2022, the reported number of new cases of the chronic form of sleeping sickness (Trypanosoma brucei gambiense) fell by 97% (from 27 862 to 799), and the number of newly reported cases of the acute form of HAT (Trypanosoma brucei rhodesiense) fell by 94% (from 619 to 38) (WHO, 2023b). These encouraging trends led the WHO to target this debilitating and highly fatal (if untreated) vector-borne parasitic disease for elimination as a public health problem by 2020, and for interruption of transmission (zero case) by 2030 (WHO, 2021, WHO, 2023a). However, the disease is persisting in many foci, and even some cases of resurgence have been documented after unfortunate events such as war or pandemics (Moore et al., 1999; Sah et al., 2023. Simarro et al). Although effective control measures, diagnosis and treatment are complex and require specific skills (WHO, 2023), especially in a context which animal reservoirs, including hidden reservoirs, can contribute to the maintenance/persistence of infection (Welburn and Maudlin, 2012; Camara et al., 2021). Vector control therefore appears as a viable alternative to accelerate sleeping sickness transmission interruption, and WHO has identified some critical actions for HAT elimination, including the coordination of vector control and animal trypanosomiasis management among countries, stakeholders and other sectors (e.g. tourism and wildlife) through multisectoral national bodies to maximize synergies (WHO, 2021). The paper by Kagbadouno and Collaborators (2024) uses microsatellite markers genotyping and population genetics tools to investigate the impact of 11 years of tiny target-based vector control on the population biology of Glossina palpalis gambiensis in Boffa, one of the three active sleeping sickness foci in Guinea (Kagbadouno et al., 2012). Although vector control significantly reduced the apparent densities of tsetse flies (and therefore the human exposure to the vector) as well as the prevalence and incidence of the disease in the Boffa HAT focus (Courtin et al., 2015), no genetic signature of vector control was observed as no difference in population size, before and after the onset of the control policy, was found. The authors then provided national programs and implementing partners with indications on the actions to be taken to (i) maintain the achievements of vector control (thus avoiding rebound/resurgence as was experienced in the past (Franco et al., 2014), and (ii) accelerate the momentum towards elimination by for example combining these vector control efforts with medical surveys for case detection and treatment, in line with WHO recommendations (WHO, 2021). References Camara M, Soumah AM, Ilboudo H, Travaillé C, Clucas C, Cooper A, Kuispond Swar NR, Camara O, Sadissou I, Calvo Alvarez E, Crouzols A, Bart JM, Jamonneau V, Camara M, MacLeod A, Bucheton B, Rotureau B. Extravascular Dermal Trypanosomes in Suspected and Confirmed Cases of gambiense Human African Trypanosomiasis. Clin Infect Dis. 2021 Jul 1;73(1):12-20. https://doi.org/10.1093/cid/ciaa897 Courtin F, Camara M, Rayaisse JB, Kagbadouno M, Dama E, Camara O, Traore IS, Rouamba J, Peylhard M, Somda MB, Leno M, Lehane MJ, Torr SJ, Solano P, Jamonneau V, Bucheton B (2015) Reducing human-tsetse contact significantly enhances the efficacy of sleeping sickness active screening campaigns: a promising result in the context of elimination. PLoS Neglected Tropical Diseases, 9. https://doi.org/10.1371/journal.pntd.0003727 Franco JR, Simarro PP, Diarra A, Jannin JG. (2014) Epidemiology of human African trypanosomiasis. Clin Epidemiol. 6:257-75. https://doi.org/10.2147/CLEP.S39728 Kagbadouno, M. S., Séré, M., Ségard, A., Camara, A. D., Camara, M., Bucheton, B., ... & Ravel, S. (2023). Population genetics of Glossina palpalis gambiensis in the sleeping sickness focus of Boffa (Guinea) before and after eight years of vector control: no effect of control despite a significant decrease of human exposure to the disease. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2023.07.25.550445 Kagbadouno MS, Camara M, Rouamba J, Rayaisse JB, Traoré IS, Camara O, Onikoyamou MF, Courtin F, Ravel S, De Meeûs T, Bucheton B, Jamonneau V, Solano P (2012) Epidemiology of sleeping sickness in boffa (Guinea): where are the trypanosomes? PLoS Neglected Tropical Diseases, 6, e1949. https://doi.org/10.1371/journal.pntd.0001949 Moore A, Richer M, Enrile M, Losio E, Roberts J, Levy D. Resurgence of sleeping sickness in Tambura County, Sudan. Am J Trop Med Hyg. 1999 Aug;61(2):315-8. https://doi.org/10.4269/ajtmh.1999.61.315 Sah R, Mohanty A, Rohilla R, Padhi BK. A resurgence of Sleeping sickness amidst the COVID-19 pandemic: Correspondence. Int J Surg Open. 2023 Apr;53:100604. https://doi.org/10.1016/j.ijso.2023.100604 Welburn SC, Maudlin I. Priorities for the elimination of sleeping sickness. Adv Parasitol. 2012;79:299-337. https://doi.org/10.1016/B978-0-12-398457-9.00004-4 World Health Organization, 2021. Ending the neglect to attain the Sustainable Development Goals: a road map for neglected tropical diseases 2021–2030. World Health Organization, Geneva, Switzerland. ISBN: 978 92 4 001035 2. 196p. World Health Organization, 2023a. Trypanosomiasis, human African (sleeping sickness): key facts. Accessed at https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) on February 19, 2023. World Health Organization, 2023b. Human African Trypanosomiasis, (sleeping sickness): the global health observatory. Accessed at https://www.who.int/data/gho/data/themes/topics/human-african-trypanosomiasis on February 19, 2023. | Population genetics of *Glossina palpalis* gambiensis in the sleeping sickness focus of Boffa (Guinea) before and after eight years of vector control: no effect of control despite a significant decrease of human exposure to the disease | Moise S. Kagbadouno, Modou Séré, Adeline Ségard, Abdoulaye Dansy Camara, Mamadou Camara, Bruno Bucheton, Jean-Mathieu Bart, Fabrice Courtin, Thierry de Meeûs, Sophie Ravel | <p style="text-align: justify;">Human African trypanosomosis (HAT), also known as sleeping sickness, is still a major concern in endemic countries. Its cyclical vector are biting insects of the genus Glossina or tsetse flies. In Guinea, the mangro... | | Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Evolution of hosts, infectious agents, or vectors, Parasites, Population genetics of hosts, infectious agents, or vectors | Hugues Nana Djeunga | 2023-07-29 13:24:52 | ||
17 Jan 2024
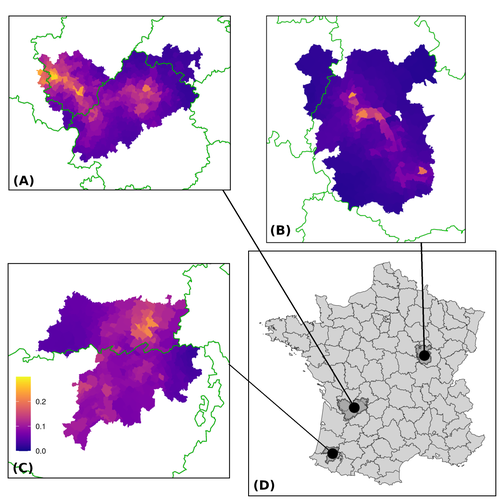
Assessing the dynamics of Mycobacterium bovis infection in three French badger populationsFrom disease surveillance to public action. Re-inforcing both epidemiological surveillance and data analysis: an illustration with Mycobacterium bovisRecommended by Jean-Francois Guégan based on reviews by Rowland Kao and 1 anonymous reviewerMycobacterium bovis, also called M. tuberculosis var. bovis, is a bacterium belonging to the M. tuberculosis complex (i.e., MTBC) and which can cause through zoonotic transmission another form of human tuberculosis (Tb). It is above all the agent of bovine tuberculosis (i.e., bTb) which affects not only cattle (wild or farmed) but also a large diversity of other wild mammals worldwide. An increasing number of infected animal cases are being discovered in many regions of the world, thus raising the problem of tuberculosis transmission, including to humans, more complex than previously thought. Efforts have been made in terms of vaccination or culling of populations of host carrier species, such as the badger for example, however leading to consequences of greater dispersion of the infectious agent. M. bovis shows a more or less significant capacity to persist outside its hosts, particularly in the environment under certain abiotic and biotic conditions. This bacillus can be transmitted and spread in many ways, including through aerosol, mucus and sputum, urine and feces, by direct contact with infected animals, their dead bodies or rather via their excreta or by inhalation of aerosols, depending on the host species concerned. In this paper, Calenge and his collaborators (Callenge et al. 2024) benefited from a national surveillance program on M. bovis cases in wild species, set up in 2011 in France, i.e., Sylvatub, for detecting and monitoring M. bovis infection in European badger (Meles meles) populations. Sylvatub is a participatory program involving both national and local stakeholder systems in order to determine changes in bTb infection levels in domestic and wild animal species. This original work had two aims: to describe spatial disease dynamics in the three clusters under scrutiny using a complex Bayesian model; and to develop indicators for the monitoring of the M. bovis infection by stakeholders and decision-makers of the program. This paper is timely and very comprehensive. In this cogent study, the authors illustrate this point by using epidemiological surveillance to obtain large amounts of data (which is generally lacking in human epidemiology, but more dramatically lacking in animal epidemiology) and a highly sophisticated biostatistical analysis (Callenge et al. 2024). It is in itself a demonstration of the current capabilities of population dynamics applied to infectious disease situations, in this case animal, in the rapidly developing discipline of disease ecology and evolution. One of the aims of the study is to propose statistical models that can be used by the different stakeholders in charge, for instance, of wildlife conservation or the regional or State veterinary services to assess disease risk in the most affected regions. References Assel AKHMETOVA, Jimena GUERRERO, Paul McADAM, Liliana CM SALVADOR, Joseph CRISPELL, John LAVERY, Eleanor PRESHO, Rowland R KAO, Roman BIEK, Fraser MENZIES, Nigel TRIMBLE, Roland HARWOOD, P Theo PEPLER, Katarina ORAVCOVA, Jordon GRAHAM, Robin SKUCE, Louis DU PLESSIS, Suzan THOMPSON, Lorraine WRIGHT, Andrew W BYRNE, Adrian R ALLEN. 2023. Genomic epidemiology of Mycobacterium bovis infection in sympatric badger and cattle populations in Northern Ireland. Microbial Genomics 9: mgen001023. https://doi.org/10.1099/mgen.0.001023 Roman BIEK, Anthony O’HARE, David WRIGHT, Tom MALLON, Carl McCORMICK, Richard J ORTON, Stanley McDOWELL, Hannah TREWBY, Robin A SKUCE, Rowland R KAO. 2012. Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathogens 8: e1003008. https://doi.org/10.1371/journal.ppat.1003008 Clément CALENGE, Ariane PAYNE, Edouard REVEILLAUD, Céline RICHOMME, Sébastien GIRARD, Stephanie DESVAUX. 2024. Assessing the dynamics of Mycobacterium bovis infection in three French badger populations. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2023.05.31.543041 Marc CHOISY, Pejman ROHANI. 2006. Harvesting can increase severity of wildlife disease epidemics. Proceedings of the Royal Society, London, Ser. B 273: 2025-2034. https://doi.org/10.1098/rspb.2006.3554 Shannon C DUFFY, Sreenidhi SRINIVASAN, Megan A SCHILLING, Tod STUBER, Sarah N DANCHUK, Joy S MICHAEL, Manigandan VENKATESAN, Nitish BANSAL, Sushila MAAN, Naresh JINDAL, Deepika CHAUDHARY, Premanshu DANDAPAT, Robab KATANI, Shubhada CHOTHE, Maroudam VEERASAMI, Suelee ROBBE-AUSTERMAN, Nicholas JULEFF, Vivek KAPUR, Marcel A BEHR. 2020. Reconsidering Mycobacterium bovis as a proxy for zoonotic tuberculosis: a molecular epidemiological surveillance study. Lancet Microbe 1: e66-e73. https://doi.org/10.1016/S2666-5247(20)30038-0 Jean-François GUEGAN. 2019. The nature of ecology of infectious disease. The Lancet Infectious Diseases 19. https://doi.org/10.1016/s1473-3099(19)30529-8 Brandon H HAYES, Timothée VERGNE, Mathieu ANDRAUD, Nicolas ROSE. 2023. Mathematical modeling at the livestock-wildlife interface: scoping review of drivers of disease transmission between species. Frontiers in Veterinary Science 10: 1225446. https://doi.org/10.3389/fvets.2023.1225446 David KING, Tim ROPER, Douglas YOUNG, Mark EJ WOOLHOUSE, Dan COLLINS, Paul WOOD. 2007. Bovine tuberculosis in cattle and badgers. Report to Secretary of State about tuberculosis in cattle and badgers. London, UK. https://www.bovinetb.info/docs/RBCT_david_%20king_report.pdf Robert MM SMITH , Francis DROBNIEWSKI, Andrea GIBSON, John DE MONTAGUE, Margaret N LOGAN, David HUNT, Glyn HEWINSON, Roland L SALMON, Brian O’NEILL. 2004. Mycobacterium bovis Infection, United Kingdom. Emerging Infectious Diseases 10: 539-541. https://doi.org/10.3201/eid1003.020819 | Assessing the dynamics of *Mycobacterium bovis* infection in three French badger populations | Clement CALENGE, Ariane PAYNE, Edouard REVEILLAUD, Celine RICHOMME, Sebastien GIRARD, Stephanie DESVAUX | <p>The Sylvatub system is a national surveillance program established in 2011 in France to monitor infections caused by <em>Mycobacterium bovis</em>, the main etiologic agent of bovine tuberculosis, in wild species. This participatory program, inv... | | Animal diseases, Ecohealth, Ecology of hosts, infectious agents, or vectors, Epidemiology, Geography of infectious diseases, Pathogenic/Symbiotic Bacteria, Zoonoses | Jean-Francois Guégan | 2023-06-05 10:50:49 | ||
16 Jul 2024
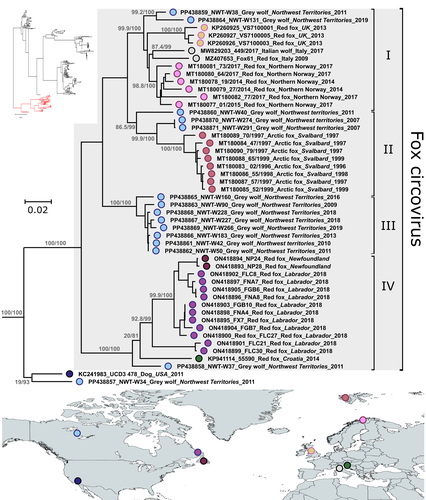
Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, CanadaWild canine viruses in the news. Better understanding multi-host transmission by adopting a disease ecology species community-based approachRecommended by Jean-Francois Guégan based on reviews by Arvind Varsani and 1 anonymous reviewerAccording to the international animal health authority, i.e., the World Organization on Animal Health (WOAH, former OIE), circoviruses are part of the Circoviridae family, which only includes 2 genera Circovirus and Cyclovirus, and infect swine, canine, ursid, viverrid, felid, pinniped, herpestid, mustelid, and several avian species (WOAH 2021). They are small (12–27 nm), non-enveloped, circular, single-stranded DNA viruses, viral replication is nuclear, and wild and domestic birds and mammals could serve as natural hosts. If most infections caused by circoviruses are subclinical in both wild and domestic species, they can be responsible for severe diseases in the commercial pig industry due to the Porcine circovirus-2 (PCV-2). These viruses can constitute a threat to wildlife, and cause their hosts to become immunocompromised, and animals often present with secondary coinfections. Canine circoviruses (CanineCV) harbour a worldwide distribution in dogs, and is the sole member of the viral genus to infect canines. They can be detected in wild carnivores, such as wolves, badgers, foxes and jackals, which indicates an ability for cross-species transmission between wildlife and domestic dogs. However, fox circovirus (FoCV), a distinct lineage of CanineCV, has been identified exclusively in wild canids (foxes and wolves) and not in dogs in Europe and North America, where it can cause in red foxes meningoencephalitis and other central nervous system signs. In their article, Canuti et al. (2024) investigate the presence, distribution and ecology of CanineCV in grey wolf specimens from the Northwest Territories, Canada. CanineCV occurrence appears to be relatively high with 45.3% positive specimens and parvoviral superinfections observed. The authors identify a high CanineCV genetic diversity among the investigated grey wolf specimens, and exacerbated by viral recombination. Phylogenetic analysis reveals the existence of 4 lineages, within each of them strains segregate by geography and not by host origin. This observed geographic segregation is interpreted as being due to the absence of exchange flows between grey wolf host subpopulations. Due to the paucity of knowledge on these circoviruses in wildlife and at the interface between wild and domestic animals, the authors discuss the plausible role of wolves as natural host reservoirs for disease transmission due to long-lasting virus-host coevolution. They are also conscious that additional maintenance hosts could exist in the wild, claiming for further studies to decipher fox circovirus disease ecology and transmission dynamics. This study underlines the importance of better understanding the transmission ecology and evolution of these Canine circoviruses, and I can only agree. Xiao et al. (2023), a research not referred to in the present work, evidenced CanineCV infection in cats in China, and obtained the first whole genome of cat-derived CanineCV. This emphasizes the importance of monitoring additional animal species and locations in the world to clarify disease ecology and transmission dynamics. A broader sampling of a wide range of animal species in different parts of the world using a species community-based approach is the key to understanding these CanineCV infections. References Marta CANUTI, Abigail V.L. KING, Giovanni FRANZO, H. Dean CLUFF, Lars E. LARSEN, Heather FENTON, Suzanne C. DUFOUR, Andrew S. LANG. 2024. Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, Canada. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2024.03.08.584028 World Organization on Animal Health. 2021. Circoviruses. https://www.woah.org/app/uploads/2021/05/circoviruses-infection-with.pdf [consulted on July 9th, 2024]. Xiangyu XIAO, Yan CHAO LI, Feng PEI XU, Xiangpi HAO, Shoujun LI, Pei ZHOU. 2023. Canine circovirus among dogs and cats in China: first identification in cats. Front. Microbiol. 14. https://doi.org/10.3389/fmicb.2023.1252272 | Diverse fox circovirus (*Circovirus canine*) variants circulate at high prevalence in grey wolves (*Canis lupus*) from the Northwest Territories, Canada | Marta Canuti, Abigail V.L. King, Giovanni Franzo, H. Dean Cluff, Lars E. Larsen, Heather Fenton, Suzanne C. Dufour, Andrew S. Lang | <p style="text-align: justify;">Canine circoviruses (CanineCV) have a worldwide distribution in dogs and are occasionally detected in wild carnivorans, indicating their ability for cross-species transmission. However, fox circovirus, a lineage of ... | | Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Epidemiology, Molecular genetics of hosts, infectious agents, or vectors, Population genetics of hosts, infectious agents, or vectors, Reservoirs, Taxonomy of hosts, infec... | Jean-Francois Guégan | Martine Peeters, Arvind Varsani | 2024-03-09 09:04:29 | |
28 May 2024
HIV self-testing positivity rate and linkage to confirmatory testing and care: a telephone survey in Côte d'Ivoire, Mali and SenegalThe benefits of HIV self-testing in West Africa: quantified.Recommended by Jessie Abbate based on reviews by 3 anonymous reviewersDespite decades of advances and understanding of the indiscriminate nature of human immunodeficiency virus (HIV), it remains shrouded in stigma that makes it difficult to reach some key populations at risk of transmission. The advent of self-testing technology for HIV (HIVST) has opened much-needed potential for bringing privacy to prevention that is crucial for curtailing its continued spread (Johnson et al., 2014). The HIV Self-Testing in Africa (STAR) Initiative (https://www.psi.org/fr/project/star/), carried out in Eastern and Southern Africa between 2015 and 2020 (Simwinga et al., 2022), demonstrated the market and public health operational potential of HIVST of different distribution methods. From 2019 to 2022, the “AutoTest de dépistage du VIH : Libre d’Accéder à la connaissance de son Statut" (ATLAS, translating to “HIVST: Freedom to know your status”) program built on these findings to quantify the public health value of HIVST for reaching key populations in West Africa (specifically, Mali, Senegal and Côte d’Ivoire) (Ky-Zerbo et al., 2022). The innovative secondary distribution methods these studies employed, where the primary targeted populations were also encouraged to take and provide tests to their contacts, helped widen the reach of HIVST within key population networks beyond those relying on access to HIV testing facilities.
The tricky part of the self-testing model lies in assessing its reach and impact while maintaining the privacy of self-testers that is central to its success. Following voluntary phone survey methods that previously were able to show expanded reach of HIVST to first-time testers in key populations in West Africa and high rates of confirmatory testing and treatment seeking (Kra et al., 2022), Kra et al. (Kra et al., 2024) quantified how many of these self-tests led to a positive result – allowing wider assessment of follow-up behaviors and positivity rates among the hard-to-reach populations the program had targeted.
While the numbers were low, the results were informative. Among respondents who reported a positive (“reactive”) HIVST, just 44% proceeded to confirmatory testing. This is lower than in other populations where HIVST follow-up has been assessed (Thirumurthy et al., 2016). The main reasons given for not confirming a reactive self-test was misinterpretation of HIVST results and not understanding that confirmatory testing was needed. The result thus highlighted a need for improved communication on how to correctly interpret HIVST results, and the authors provided ranges for how this misinterpretation could have affected their positivity estimates. However, the majority of those who sought confirmatory testing did so within 3 months, and nearly all of those with confirmed infection started on treatment. HIV positivity rates in the three countries were all higher than other published HIV positivity estimates (Giguère et al., 2021; Maheu-Giroux et al., 2019), suggesting that HIVST methods were highly effective at reaching the targeted communities. Finally, while the authors demonstrated their methods as an effective way of assessing the utility of HIVST campaigns and identifying ways to improve them, the follow-up surveys are likely too costly to replace current passive surveillance methods for assessing community disease burden. That said, these precious data should be taken as validation of the public health value of HIV self-testing in key populations across communities in West Africa. With improvements in communicating instructions for use and follow-up, there is little doubt that the innovation of HIVST primary and secondary distribution could become a widely useful addition to the fight against HIV.
References Giguère, K., Eaton, J. W., Marsh, K., Johnson, L. F., Johnson, C. C., Ehui, E., Jahn, A., Wanyeki, I., Mbofana, F., Bakiono, F., Mahy, M., & Maheu-Giroux, M. (2021). Trends in knowledge of HIV status and efficiency of HIV testing services in sub-Saharan Africa, 2000–20: a modelling study using survey and HIV testing programme data. The Lancet HIV, 8(5), e284–e293. https://doi.org/10.1016/S2352-3018(20)30315-5 Johnson, C., Baggaley, R., Forsythe, S., Van Rooyen, H., Ford, N., Napierala Mavedzenge, S., Corbett, E., Natarajan, P., & Taegtmeyer, M. (2014). Realizing the potential for HIV self-testing. In AIDS and Behavior (Vol. 18, Issue SUPPL. 4). Springer New York LLC. https://doi.org/10.1007/s10461-014-0832-x Kra, A. K., Fosto, A. S., N’guessan, K. N., Geoffroy, O., Younoussa, S., Kabemba, O. K., Gueye, P. A., Ndeye, P. D., Rouveau, N., Boily, M. C., Silhol, R., d’Elbée, M., Maheu-Giroux, M., Vautier, A., & Larmarange, J. (2022). Can HIV self-testing reach first-time testers? A telephone survey among self-test end users in Côte d’Ivoire, Mali, and Senegal. BMC Infectious Diseases, 22. https://doi.org/10.1186/s12879-023-08626-w Kra, A. K., Fotso, A. S., Rouveau, N., Maheu-Giroux, M., Boily, M.-C., Silhol, R., d’Elbée, M., Vautier, A., Lamarange, J., & the Atlas team. (2024). HIV self-testing positivity rate and linkage to confirmatory testing and care: a telephone survey in Côte d’Ivoire, Mali, and Senegal. MedRxiv, Ver. 4 Peer-Reviewed and Recommended by Peer Community in Infections, 2023.06.10.23291206. https://doi.org/https://doi.org/10.1101/2023.06.10.23291206 Ky-Zerbo, O., Desclaux, A., Boye, S., Maheu-Giroux, M., Rouveau, N., Vautier, A., Camara, C. S., Kouadio, B. A., Sow, S., Doumenc-Aidara, C., Gueye, P. A., Geoffroy, O., Kamemba, O. K., Ehui, E., Ndour, C. T., Keita, A., & Larmarange, J. (2022). “I take it and give it to my partners who will give it to their partners”: Secondary distribution of HIV self-tests by key populations in Côte d’Ivoire, Mali, and Senegal. BMC Infectious Diseases, 22. https://doi.org/10.1186/s12879-023-08319-4 Maheu-Giroux, M., Marsh, K., Doyle, C. M., Godin, A., Lanièce Delaunay, C., Johnson, L. F., Jahn, A., Abo, K., Mbofana, F., Boily, M. C., Buckeridge, D. L., Hankins, C. A., & Eaton, J. W. (2019). National HIV testing and diagnosis coverage in sub-Saharan Africa: A new modeling tool for estimating the “first 90” from program and survey data. AIDS, 33, S255–S269. https://doi.org/10.1097/QAD.0000000000002386 Simwinga, M., Gwanu, L., Hensen, B., Sigande, L., Mainga, M., Phiri, T., Mwanza, E., Kabumbu, M., Mulubwa, C., Mwenge, L., Bwalya, C., Kumwenda, M., Mubanga, E., Mee, P., Johnson, C. C., Corbett, E. L., Hatzold, K., Neuman, M., Ayles, H., & Taegtmeyer, M. (2022). Lessons learned from implementation of four HIV self-testing (HIVST) distribution models in Zambia: applying the Consolidated Framework for Implementation Research to understand impact of contextual factors on implementation. BMC Infectious Diseases, 22(Suppl 1). https://doi.org/10.1186/s12879-024-09168-5 Thirumurthy, H., Masters, S. H., Mavedzenge, S. N., Maman, S., Omanga, E., & Agot, K. (2016). Promoting male partner HIV testing and safer sexual decision making through secondary distribution of self-tests by HIV-negative female sex workers and women receiving antenatal and post-partum care in Kenya: a cohort study. The Lancet HIV, 3(6), e266–e274. https://doi.org/10.1016/S2352-3018(16)00041-2
| HIV self-testing positivity rate and linkage to confirmatory testing and care: a telephone survey in Côte d'Ivoire, Mali and Senegal | Kra Djuhe Arsene Kouassi, Arlette Simo Fotso, Nicolas Rouveau, Mathieu Maheu-Giroux, Marie-Claude Boily, Romain Silhol, Marc d'Elbee, Anthony Vautier, Joseph Larmarange, ATLAS Team | <p>HIV self-testing (HIVST) empowers individuals to decide when and where to test and with whom to share their results. From 2019 to 2022, the ATLAS program distributed ~ 400 000 HIVST kits in Côte d’Ivoire, Mali, and Senegal. It prioritised key p... | | Epidemiology | Jessie Abbate | 2023-06-16 16:40:51 | ||
21 Jul 2022

Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV)Understanding the in vitro evolution of Cyprinid herpesvirus 3 (CyHV-3), a story of structural variations that can lead to the design of attenuated virus vaccinesRecommended by Jorge Amich based on reviews by Lucie Cappuccio and Veronique Hourdel
Structural variations (SVs) play a key role in viral evolution, and therefore they are also important for infection dynamics. However, the contribution of structural variations to the evolution of double-stranded viruses is limited. This knowledge can help to understand the population dynamics and might be crucial for the future development of viral attenuated vaccines. In this study, Fuandila et al (1) use the Cyprinid herpesvirus 3 (CyHV-3), commonly known as koi herpesvirus (KHV), to investigate the variability and contribution of structural variations (SV) for viral evolution after 99 passages in vitro. This virus, with the largest genome among herperviruses, causes a lethal infection in common carp and koi associated with mortalities up to 95% (2). Interestingly, KHV infections are caused by haplotype mixtures, which possibly are a source of genome diversification, but make genomic comparisons more difficult. The authors have used ultra-deep long-read sequencing of two passages, P78 and P99, which were previously described to have differences in virulence. They have found a surprisingly high and wide distribution of SVs along the genome, which were enriched in inversion and deletion events and that often led to defective viral genomes. Although it is known that these defective viral genomes negatively impact viral replication, their implications for virus persistence are still unclear. Subsequently, the authors concentrated on the virulence-relevant region ORF150, which was found to be different in P78 (deletion in 100% of the reads) and P99 (reference-like haplotype). To understand this loss and gain of full ORF150, they searched for SV turn-over in 10 intermediate passages. This analysis revealed that by passage 10 deleted and inverted (attenuated) haplotypes had already appeared, steadily increased frequency until P78, and then completely disappeared between P78 and P99. This is a striking result that raises new questions as to how this clearance occurs, which is really important as these reversions may result in undesirable increases in virulence of live-attenuated vaccines. We recommend this preprint because its use of ultra-deep long-read sequencing has permitted to better understand the role of SV diversity and dynamics in viral evolution. This study shows an unexpectedly high number of structural variations, revealing a novel source of virus diversification and confirming the different mixtures of haplotypes in different passages, including the gain of function. This research provides basic knowledge for the future design of live-attenuated vaccines, to prevent the reversion to virulent viruses. References (1) Fuandila NN, Gosselin-Grenet A-S, Tilak M-K, Bergmann SM, Escoubas J-M, Klafack S, Lusiastuti AM, Yuhana M, Fiston-Lavier A-S, Avarre J-C, Cherif E (2022) Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV). bioRxiv, 2022.03.10.483410, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.03.10.483410 (2) Sunarto A, McColl KA, Crane MStJ, Sumiati T, Hyatt AD, Barnes AC, Walker PJ. Isolation and characterization of koi herpesvirus (KHV) from Indonesia: identification of a new genetic lineage. Journal of Fish Diseases, 34, 87-101. https://doi.org/10.1111/j.1365-2761.2010.01216.x | Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV) | Nurul Novelia Fuandila, Anne-Sophie Gosselin-Grenet, Marie-Ka Tilak, Sven M Bergmann, Jean-Michel Escoubas, Sandro Klafack, Angela Mariana Lusiastuti, Munti Yuhana, Anna-Sophie Fiston-Lavier, Jean-Christophe Avarre, Emira Cherif | <p style="text-align: justify;">Structural variations (SVs) constitute a significant source of genetic variability in virus genomes. Yet knowledge about SV variability and contribution to the evolutionary process in large double-stranded (ds)DNA v... | | Animal diseases, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Viruses | Jorge Amich | Lucie Cappuccio, Veronique Hourdel | 2022-03-11 10:50:50 | |
07 Oct 2022

Guidelines for the reliable use of high throughput sequencing technologies to detect plant pathogens and pestsHigh-throughput sequencing for the diagnostic of plant pathologies and identification of pests: recommendations and challengesRecommended by Olivier Schumpp based on reviews by Denise Altenbach and David RoquisHigh-throughput sequencing (HTS) has revealed an incredible diversity of microorganisms in ecosystems and is also changing the monitoring of macroorganism biodiversity (Deiner et al. 2017; Piper et al. 2019). The diagnostic of plant pathogens and the identification of pests is gradually integrating the use of these techniques, but there are still obstacles. Most of them are related to the reliability of these analyses, which have long been considered insufficient because of their dependence on a succession of sophisticated operations involving parameters that are sometimes difficult to adapt to complex matrices or certain diagnostic contexts. The need to validate HTS approaches is gradually being highlighted in recent work but remains poorly documented (Bester et al. 2022). In this paper, a large community of experts presents and discusses the key steps for optimal control of HTS performance and reliability in a diagnostic context (Massart et al. 2022). It also addresses the issue of costs. The article provides recommendations that closely combine the quality control requirements commonly used in conventional diagnostics with newer or HTS-specific control elements and concepts that are not yet widely used. It discusses the value of these for the use of the various techniques currently covered by the terms "High Throughput Sequencing" in diagnostic activities. The elements presented are intended to limit false positive or false negative results but will also optimise the interpretation of contentious results close to the limits of analytical sensitivity or unexpected results, both of which appear to be frequent when using HTS. Furthermore, the need for risk analysis, verification and validation of methods is well illustrated with numerous examples for each of the steps considered crucial to ensure reliable use of HTS. The clear contextualisation of the proposals made by the authors complements and clarifies the need for user expertise according to the experimental objectives. Some unanswered questions that will require further development and validation are also presented. This article should benefit a large audience including researchers with some level of expertise in HTS but unfamiliar with the recent concepts of controls common in the diagnostic world as well as scientists with strong diagnostic expertise but less at ease with the numerous and complex procedures associated with HTS. References Bester R, Steyn C, Breytenbach JHJ, de Bruyn R, Cook G, Maree HJ (2022) Reproducibility and Sensitivity of High-Throughput Sequencing (HTS)-Based Detection of Citrus Tristeza Virus and Three Citrus Viroids. Plants, 11, 1939. https://doi.org/10.3390/plants11151939 Deiner K, Bik HM, Mächler E, Seymour M, Lacoursière-Roussel A, Altermatt F, Creer S, Bista I, Lodge DM, de Vere N, Pfrender ME, Bernatchez L (2017) Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Molecular Ecology, 26, 5872–5895. https://doi.org/10.1111/mec.14350 Massart, S et al. (2022) Guidelines for the reliable use of high throughput sequencing technologies to detect plant pathogens and pests. Zenodo, 6637519, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.5281/zenodo.6637519 Piper AM, Batovska J, Cogan NOI, Weiss J, Cunningham JP, Rodoni BC, Blacket MJ (2019) Prospects and challenges of implementing DNA metabarcoding for high-throughput insect surveillance. GigaScience, 8, giz092. https://doi.org/10.1093/gigascience/giz092 | Guidelines for the reliable use of high throughput sequencing technologies to detect plant pathogens and pests | S. Massart, I. Adams, M. Al Rwahnih, S. Baeyen, G. J. Bilodeau, A. G. Blouin, N. Boonham, T. Candresse, A. Chandelier, K. De Jonghe, A. Fox, Y.Z.A. Gaafar, P. Gentit, A. Haegeman, W. Ho, O. Hurtado-Gonzales, W. Jonkers, J. Kreuze, D. Kutjnak, B. B... | <p style="text-align: justify;">High-throughput sequencing (HTS) technologies have the potential to become one of the most significant advances in molecular diagnostics. Their use by researchers to detect and characterize plant pathogens and pests... | | Diagnosis, Pest management, Phytopathology, Plant diseases | Olivier Schumpp | 2022-06-13 11:26:18 | ||
06 Apr 2023

Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populationsChanges in aggressiveness in pathotypes of wheat leaf rustRecommended by Pierre Gladieux based on reviews by 2 anonymous reviewersUnderstanding the ecological and evolutionary factors underlying the spread of new fungal pathogen populations can inform the development of more effective management strategies. In plant pathology, pathogenicity is generally presented as having two components: ‘virulence’ (qualitative pathogenicity) and aggressiveness (quantitative pathogenicity). Changes in virulence in response to the deployment of new resistant varieties are a major driver of the spread of new populations (called pathotypes, or races) in modern agrosystems, and the genomic (i.e. proximal) and eco-evolutionary (i.e. ultimate) factors underlying these changes are well-documented [1,2,3]. By contrast, the role of changes in aggressiveness in the spread of pathotypes remains little known [4]. The study by Cécilia Fontyn and collaborators [5] set out to characterize changes in aggressiveness for isolates of two pathotypes of the wheat leaf rust (Puccinia triticina) that have been dominant in France during the 2005-2016 period. Isolates were genetically characterized using multilocus microsatellite typing and phenotypically characterized for three components of aggressiveness on wheat varieties: infection efficiency, latency period, and sporulation capacity. Using experiments that represent quite a remarkable amount of work and effort, Fontyn et al. showed that each dominant pathotype consisted of several genotypes, including common genotypes whose frequency changed over time. For each pathotype, the genotypes that were more common initially were replaced by a more aggressive genotype. Together, these results show that changes in the genetic composition of populations of fungal plant pathogens can be associated with, and may be caused by, changes in the quantitative components of pathogenicity. This study also illustrates how extensive, decade-long monitoring of fungal pathogen populations, such as the one conducted for wheat leaf rust in France, represents a very valuable resource for research. REFERENCES [1] Brown, J. K. (1994). Chance and selection in the evolution of barley mildew. Trends in Microbiology, 2(12), 470-475. https://doi.org/10.1016/0966-842x(94)90650-5 [2] Daverdin, G., Rouxel, T., Gout, L., Aubertot, J. N., Fudal, I., Meyer, M., Parlange, F., Carpezat, J., & Balesdent, M. H. (2012). Genome structure and reproductive behaviour influence the evolutionary potential of a fungal phytopathogen. PLoS Pathogens, 8(11), e1003020. https://doi.org/10.1371/journal.ppat.1003020 [3] Gladieux, P., Feurtey, A., Hood, M. E., Snirc, A., Clavel, J., Dutech, C., Roy, M., & Giraud, T. (2015). The population biology of fungal invasions.Molecular Ecology, 24(9), 1969-86. https://doi.org/10.1111/mec.13028 [4] Fontyn, C., Zippert, A. C., Delestre, G., Marcel, T. C., Suffert, F., & Goyeau, H. (2022). Is virulence phenotype evolution driven exclusively by Lr gene deployment in French Puccinia triticina populations?. Plant Pathology, 71(7), 1511-1524. https://doi.org/10.1111/ppa.13599 [5] Fontyn, C., Meyer, K. J., Boixel, A. L., Delestre, G., Piaget, E., Picard, C., Suffer, F., Marcel, T.C., & Goyeau, H. (2022). Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations. bioRxiv, 2022.08.29.505401, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.29.505401 | Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations | Cécilia FONTYN, Kevin JG MEYER, Anne-Lise BOIXEL, Ghislain DELESTRE, Emma PIAGET, Corentin PICARD, Frédéric SUFFERT, Thierry C MARCEL, Henriette GOYEAU | <p style="text-align: justify;">Plant pathogens are constantly evolving and adapting to their environment, including their host. Virulence alleles emerge, and then increase, and sometimes decrease in frequency within pathogen populations in respon... | | Coevolution, Epidemiology, Evolution of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Pathogenic/Symbiotic Fungi, Phytopathology, Plant diseases, Population dynamics of hosts, infectious agents, or... | Pierre Gladieux | Emerson Del Ponte , Jacqui Shykoff, Leïla Bagny Beilhe , Alexey Mikaberidze | 2022-09-29 20:01:57 | |
08 Aug 2023
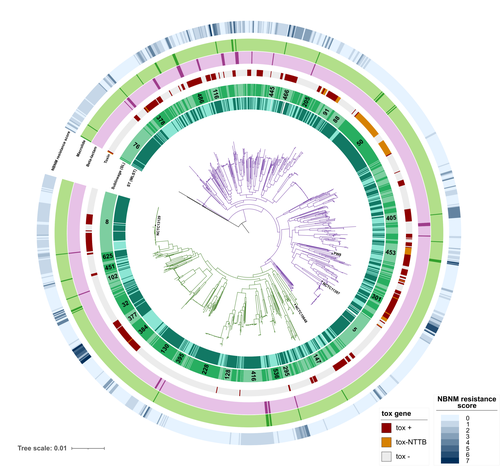
A global Corynebacterium diphtheriae genomic framework sheds light on current diphtheria reemergenceDIPHTOSCAN : A new tool for the genomic surveillance of diphtheriaRecommended by Rodolfo García-Contreras based on reviews by Ankur Mutreja and 2 anonymous reviewersOne of the greatest achievements of health sciences is the eradication of infectious diseases such as smallpox that in the past imposed a severe burden on humankind, through global vaccination campaigns. Moreover, progress towards the eradication of others such as poliomyelitis, dracunculiasis, and yaws is being made. In contrast, other infections that were previously contained are reemerging, due to several factors, including lack of access to vaccines due to geopolitical reasons, the rise of anti-vaccine movements, and the constant mobility of infected persons from the endemic sites. One of such disease is diphtheria, caused by Corynebacterium diphtheriae and a few other related species such as C. ulcerans and C. pseudotuberculosis. Importantly, in France, diphtheria cases reported in 2022 increased 7-fold from the average of previously recorded cases per year in the previous 4 years and the situation in other European countries is similar. Hence, as reported here, Hennart et al. (2023) developed DIPHTOSCAN, a free access bioinformatics tool with user-friendly interphase, aimed to easily identify, extract and interpret important genomic features such as the sublineage of the strain, the presence of the tox gene (as a string predictor for toxigenic disease) as well as genes coding other virulence factors such as fimbriae, and the presence of know resistant mechanisms towards antibiotics like penicillin and erythromycin currently used in the clinic to treat this infection. The authors validated the performance of their tool with a large collection of genomes, including those obtained from the isolates of the 2022 outbreak in France, more than 1,200 other genomes isolated from France, Algeria, and Yemen, and more than 500 genomes from several countries from Europe, America, Africa, Asia, and Oceania that are available through the NCBI site. DIPHTOSCAN will allow the rapid identification and surveillance of potentially dangerous strains such as those being tox-positive isolates and resistant to multiple drugs and/or first-line treatments and a better understanding of the epidemiology and evolution of this important reemerging disease. | A global *Corynebacterium diphtheriae* genomic framework sheds light on current diphtheria reemergence | Melanie Hennart, Chiara Crestani, Sebastien Bridel, Nathalie Armatys, Sylvie Brémont, Annick Carmi-Leroy, Annie Landier, Virginie Passet, Laure Fonteneau, Sophie Vaux, Julie Toubiana, Edgar Badell, Sylvain Brisse | <p style="text-align: justify;"><strong>Background</strong></p> <p style="text-align: justify;">Diphtheria, caused by <em>Corynebacterium diphtheriae</em>, reemerges in Europe since 2022. Genomic sequencing can inform on transmission routes and g... | | Drug resistance, tolerance and persistence, Epidemiology, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Microbiology of infections, Population genetics of hosts, infectiou... | Rodolfo García-Contreras | Ankur Mutreja | 2023-03-09 16:02:27 | |
21 Sep 2023
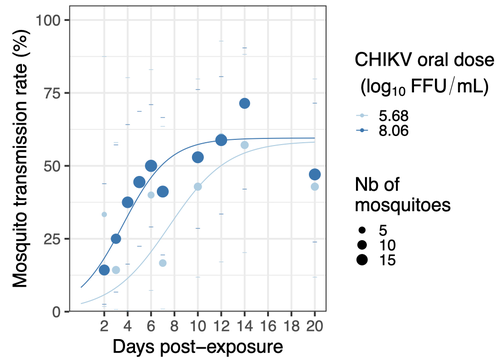
Chikungunya intra-vector dynamics in Aedes albopictus from Lyon (France) upon exposure to a human viremia-like dose range reveals vector barrier permissiveness and supports local epidemic potentialFill in one gap in our understanding of CHIKV intra-vector dynamicsRecommended by Sara Moutailler based on reviews by 2 anonymous reviewers
Mosquitoes are first vector of pathogen worldwide and transmit several arbovirus, most of them leading to major outbreaks (1). Chikungunya virus (CHIKV) is a perfect example of the “explosive type” of arbovirus, as observed in La Réunion Island in 2005-2006 (2-6) and also in the outbreak of 2007 in Italy (7), both vectorized by Ae. albopictus. Being able to better understand CHIKV intra-vector dynamics is still of major interest since not all chikungunya strain are explosive ones (8). In this study (9), the authors have evaluated the vector competence of a local strain of Aedes albopictus (collected in Lyon, France) for CHIKV. They evaluated infection, dissemination and transmission dynamics of CHIKV using different dose of virus in individual mosquitoes from day 2 to day 20 post exposure, by titration and quantification of CHIKV RNA load in the saliva. As highlighted by both reviewers, the most innovative idea in this study was the use of three different oral doses trying to span human viraemia detected in two published studies (10-11), doses that were estimated through their model of human CHIKV viremia in the blood. They have found that CHIKV dissemination from the Ae. albopictus midgut depends on the interaction between time post-exposure and virus dose (already highlighted by other international publications). Then their results were implemented in the agent-based model nosoi to estimate the epidemic potential of CHIKV in a French population of Ae. albopictus, using realistic vectorial capacity parameters. To conclude, the authors have discussed the importance of other parameters that could influence vector competence as mosquito microbiota and temperature, parameters that need also to be estimated in local mosquito population to improve the risk assessment through modelling. As pointed out by both reviewers, this is a nice study, well written and easy to read. These results allow filling in another gap of our understanding of CHIKV intra-vector dynamics and highlight the epidemic potential of CHIKV upon transmission by Aedes albopictus in mainland France. For all these reasons, I chose to recommend this article for Peer Community In Infections. References 1. Marine Viglietta, Rachel Bellone, Adrien Albert Blisnick, Anna-Bella Failloux. (2021). Vector Specificity of Arbovirus Transmission. Front Microbiol Dec 9;12:773211. https://doi.org/10.3389/fmicb.2021.773211 2. Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney M-C, Lavenir R, Pardigon N, Reynes J-M, Pettinelli F, Biscornet L, Diancourt L, Michel S, Duquerroy S, Guigon G, Frenkiel M-P, Bréhin A-C, Cubito N, Desprès P, Kunst F, Rey FA, Zeller H, Brisse S. (2006). Genome Microevolution of Chikungunya viruses Causing the Indian Ocean Outbreak. 2006. PLoS Medicine, 3, e263. https://doi.org/10.1371/journal.pmed.0030263 3. Bonilauri P, Bellini R, Calzolari M, Angelini R, Venturi L, Fallacara F, Cordioli P, 687 Angelini P, Venturelli C, Merialdi G, Dottori M. (2008). Chikungunya Virus in Aedes albopictus, Italy. Emerging Infectious 689 Diseases, 14, 852–854. https://doi.org/10.3201/eid1405.071144 4. Pagès F, Peyrefitte CN, Mve MT, Jarjaval F, Brisse S, Iteman I, Gravier P, Tolou H, Nkoghe D, Grandadam M. (2009). Aedes albopictus Mosquito: The Main Vector of the 2007 Chikungunya Outbreak in Gabon. PLoS ONE, 4, e4691. https://doi.org/10.1371/journal.pone.0004691 5. Paupy C, Kassa FK, Caron M, Nkoghé D, Leroy EM (2012) A Chikungunya Outbreak Associated with the Vector Aedes albopictus in Remote Villages of Gabon. Vector-Borne and Zoonotic Diseases, 12, 167–169. https://doi.org/10.1089/vbz.2011.0736 6. Mombouli J-V, Bitsindou P, Elion DOA, Grolla A, Feldmann H, Niama FR, Parra H-J, Munster VJ. (2013). Chikungunya Virus Infection, Brazzaville, Republic of Congo, 2011. Emerging Infectious Diseases, 19, 1542–1543. https://doi.org/10.3201/eid1909.130451 7. Venturi G, Luca MD, Fortuna C, Remoli ME, Riccardo F, Severini F, Toma L, Manso MD, Benedetti E, Caporali MG, Amendola A, Fiorentini C, Liberato CD, Giammattei R, Romi R, Pezzotti P, Rezza G, Rizzo C. (2017). Detection of a chikungunya outbreak in Central Italy, August to September 2017. Eurosurveillance, 22, 17–00646. https://doi.org/10.2807/1560-7917.es.2017.22.39.17-00646 8. de Lima Cavalcanti, T.Y.V.; Pereira, M.R.; de Paula, S.O.; Franca, R.F.d.O. (2022). A Review on Chikungunya Virus Epidemiology, Pathogenesis and Current Vaccine Development. Viruses 2022, 14, 969. https://doi.org/10.3390/v14050969 9. Barbara Viginier, Lucie Cappuccio, Celine Garnier, Edwige Martin, Carine Maisse, Claire Valiente Moro, Guillaume Minard, Albin Fontaine, Sebastian Lequime, Maxime Ratinier, Frederick Arnaud, Vincent Raquin. (2023). Chikungunya intra-vector dynamics in Aedes albopictus from Lyon (France) upon exposure to a human viremia-like dose range reveals vector barrier permissiveness and supports local epidemic potential. medRxiv, ver.3, peer-reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2022.11.06.22281997 10. Appassakij H, Khuntikij P, Kemapunmanus M, Wutthanarungsan R, Silpapojakul K (2013) Viremic profiles in CHIKV-infected cases. Transfusion, 53, 2567–2574. https://doi.org/10.1111/j.1537-2995.2012.03960.x 11. Riswari SF, Ma’roef CN, Djauhari H, Kosasih H, Perkasa A, Yudhaputri FA, Artika IM, Williams M, Ven A van der, Myint KS, Alisjahbana B, Ledermann JP, Powers AM, Jaya UA (2015) Study of viremic profile in febrile specimens of chikungunya in Bandung, Indonesia. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology, 74, 61–5. https://doi.org/10.1016/j.jcv.2015.11.017 | Chikungunya intra-vector dynamics in *Aedes albopictus* from Lyon (France) upon exposure to a human viremia-like dose range reveals vector barrier permissiveness and supports local epidemic potential | Barbara Viginier, Lucie Cappuccio, Celine Garnier, Edwige Martin, Carine Maisse, Claire Valiente Moro, Guillaume Minard, Albin Fontaine, Sebastian Lequime, Maxime Ratinier, Frederick Arnaud, Vincent Raquin | <p>Arbovirus emergence and epidemic potential, as approximated by the vectorial capacity formula, depends on host and vector parameters, including the vector intrinsic ability to replicate then transmit the pathogen known as vector competence. Vec... | | Epidemiology, Vectors, Viruses | Sara Moutailler | 2023-06-17 15:59:17 |




