
Recommendation
Deciphering the virome (the set or assemblage of viruses) of the Earth, from individual organisms to entire ecosystems, has become a key priority. The first step to better understanding the impact of viruses on the ecology and functions of ecosystems is to describe their diversity. Such knowledge opens the gates to a better assessment of global nutrient cycling or of the threat that viruses represent to individual health. This explains the increasing number of pioneering studies that are currently sequencing the complete or partial genome of thousands of new viruses [1].
In their exciting study, Fritz and collaborators [2], authors sampled 209 army ants (Genus Dorylus) to investigate the virus diversity in dense forests that researchers cannot easily access. Indeed, these ants live in colonies (21 were sampled) that can move 1 km per day, covering a significant area and attacking many invertebrate and vertebrate preys. Each sample was sequenced by a protocol called VANA sequencing and allowing the enrichment of the sample in viral sequences [3], so improving the detection of viruses present at low abundance in the ant (and more specifically in its gut for viruses infecting preys).
Around 45,000 contigs presented homologies with bacterial, plant, invertebrate, and vertebrate infecting viruses. Half could be assigned to 56 families and 157 genera of the International Committee on Taxonomy of Viruses. Beyond this amazing harvest of new and known virus sequences using an original methodology, the results significantly improve the current frontiers of known viral taxonomy and diversity and raise exciting research tracks to expand them.
As a preprint, several blogs or news of leading scientists and journals have already highlighted this study. For example, in the news section of Science magazine, Jon Cohen underlined the originality of the approach for virus hunting on Earth with the title “Armed with air samplers, rope tricks, and—yes—ants, virus hunters spot threats in new ways”[4]. Another example is the mention of the publication by Elisabeth Bik in her Microbiome Digest: she wrote, “An amazing read is a fresh preprint from Fritz and collaborator describing an exciting method of sampling in difficult-to-reach environments“ [5].
The paper from Fritz et al [2] thus represents a significant advance in virus ecology, as already recognized by early readers, and this is why I strongly recommend its publication in PCI Infections.
REFERENCES
1. Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, Lohr D, Novakovsky G, Buchfink B, Al-Shayeb B, Banfield JF, de la Peña M, Korobeynikov A, Chikhi R, Babaian A (2022) Petabase-scale sequence alignment catalyses viral discovery. Nature, 602, 142–147. https://doi.org/10.1038/s41586-021-04332-2
2. Fritz M, Reggiardo B, Filloux D, Claude L, Fernandez E, Mahé F, Kraberger S, Custer JM, Becquart P, Mebaley TN, Kombila LB, Lenguiya LH, Boundenga L, Mombo IM, Maganga GD, Niama FR, Koumba J-S, Ogliastro M, Yvon M, Martin DP, Blanc S, Varsani A, Leroy E, Roumagnac P (2023) African army ants at the forefront of virome surveillance in a remote tropical forest. bioRxiv, 2022.12.13.520061, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.12.13.520061
3. François S, Filloux D, Fernandez E, Ogliastro M, Roumagnac P (2018) Viral Metagenomics Approaches for High-Resolution Screening of Multiplexed Arthropod and Plant Viral Communities. In: Viral Metagenomics: Methods and Protocols Methods in Molecular Biology. (eds Pantaleo V, Chiumenti M), pp. 77–95. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7683-6_7
4. Cohen J (2023) Virus hunters test new surveillance tools. Science, 379, 16–17. https://doi.org/10.1126/science.adg5292
5. Ponsero A (2023) February 18th, 2023. Microbiome Digest - Bik’s Picks. https://microbiomedigest.com/2023/02/18/february-18th-2023/
DOI or URL of the preprint: https://doi.org/10.1101/2022.12.13.520061
Version of the preprint: 1
Dear authors,
On behalf of the board of Peer Community in Infection, I would like to thank you for considering it for sending your publication.
I am please to send you the decision related to the publication sent to Peer Community In Infection whose title is "African army ants at the forefront of virome surveillance in a remote tropical forest". The publication is accepted in PCI Infection and we propose several improvements in the next sections.
The document has been reviewed positively by both reviewers and I have also added some suggestions after reading carefully the manuscript. We have all appreciated the originality of the work and the clarity of the document which is well written taking into account the huge amount of results and discoveries from this innovative approach.
You can find the comments and suggestions of both reviewers and myself in this response and we thank you in advance for answering these point by point while adapting the text when necessary.
Comments from the recommender:
1. Introduction
- 87 millions of eukaryotic virus species on earth
This number is an estimation of the number of viral species per eukaryotic species multiplied by the estimation of eukaryotic species on earth. This double estimation has a very large uncertainty and I suggest to eliminate the number but simply indicating there are millions of viruses which already illustrates the gaps with ICTV recognized species
- The organisms that we farm
Thamed organisms, being animals or plants could better represent the borders as dogs or cats are not farmed for example
2. Material and methods
- Positive and negative controls have been used, which is very positive and can rise the confidence in the obtained results but what were the positive and negative controls used ?
- The minimal length of the contig is well justified but the e-value threshold not. Can it be explained ? Indeed, there might be a risk of detecting Enogenous Viral Elements (EVEs) from genome sequences of the hosts with such value.
- How can the link be done between the supplementary table 1 (identifying each sample) and the raw data presented in SRA, more specifically the internal tags identifying each sample within a library (e.g. the 3 pooled sequencing dataset MGN-1, MGN-2 and MGN-3 by Illumina and Flongle sequencing)) ? I could not find it. So adding a column in Supplementary table 1 with the corresponding tags used would facilitate reanalysis of the data of this pioneering sequencing effort by the scientific community
3. Results
- There is no information on the results of the controls and how it helped in results interpretation (as it has been considered as the third step of bioinformatic analysis in a recent publication – DOI: 10.24072/pcjournal.181 - and in a new EPPO standard PM7/151.
- This point is related to reviewer 1 comment concerning the ~24,000 contigs potentially of viral origin but without similarity to viral genera recognized by ICTV: what to do with them ? They are not really discussed while they could have a great interest in filling the knowledge gaps between existing viruses on earth and already discovered ones (although I acknowledge it is important to remain cautious about them and not being too speculative).
- Were PeVD and SoMV the only known plant viruses detected ?
- While using VANA, how do you explain the small contigs retrieved from plant viruses ? It means complete viral particles were not recovered or the sequencing depth was not high enough as the initial concentrations of the plant viruses were too low ? This could be discussed (maybe more broadly for viruses infecting non-arthropod hosts)
Basic reporting
This paper provides an insightful analysis of a virome surveillance. It offers an original approach that is both rigorous and accessible. The findings are mostly well-supported, and it is likely to be highly cited. The paper is an elegant example of research that will be widely recognized.
This article presents an original approach to the virome of a remote ecosystem by using ants as a proxy. With just 209 ants, the authors were able to detect 22,406 virus-like contigs belonging to 56 families. Seventeen of the 29 ant colonies were identified thanks to the “accidental/non targeted” recovery of the COI gene. This approach is likely to lead to an increased level of detection in poorly studied areas. This will be beneficial for the global virome description. Notably, the authors highlighted the overrepresented families of Parvoviridae and Circoviridae. Sequences of 403 Parvovirus were analysed based on their SF3 proteins, with more than 200 amino acids available for comparison with publicly available data. This revealed an increased diversity, as well as an expanded geographical distribution and potential host range. Additionally, 45 complete genomes of novel cyclovirus were resequenced and compared with publicly available data, providing further insights into this family.
Experimental design
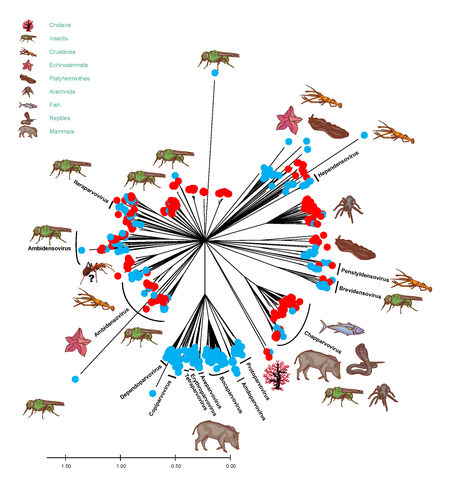
The work on the Parvoviridae and Circoviridae families is very thourough; however, due to the nature of the sequences, similar conclusions could not be made for other virus groups. The number of contigs and virus families identified in the study were based on contigs with lengths ≥200 nt, and retained viral BLASTx assignations of these contigs with e-values < 0.001 (M&M). It is not specified in the text that the BlastX was done on a complete non-redundant protein database (GenBank non-redundant database is indicated on the legend of fig 1). The amino acid identity recovered, as reported in Figure 1, was as low as <25%. Figure 1 is informative but can be misleading as a virus species can be represented multiple times, e.g. the two closely related points for the nepovirus can represent two different viruses or two contigs covering different parts of this segmented virus. In addition, the percentage of homology represented in figure 1 can be from very conserved genes (e.g. RdRP) or from putative genes with low homology even within well described families (the same virus could have multiple contigs with very varied homology to the closest sequence from the database). The legend of this figure should also be clarified as to whether the amino acids homology is per sequence alignment, or the homology given by BlastX, where only the matching region of the molecule is measured (in which case, this can be a fraction of a short 200 nt contigs (67 aa)).
In the manuscript, the authors have been cautious not to overstate their findings. It is evident that ants are a good proxy to access difficult regions, and the authors note that the ants are “not completely unbiased”. Judging by Figure 1, they are clearly biased towards animal, mostly invertebrate ssDNA viruses (as mentioned p14L12). Few plant viruses are detected and mycoviruses are not discussed at all. The fact that these viruses have to pass through additional steps in the trophic chain is discussed on page 19, but what can be said about viruses with low stability, concentration, or prevalence? The principle of VANA should yield nucleic acids protected by a capsid (in contradiction with the degradation observed). Are ants the best candidates for a plant metavirome? The authors should provide a more detailed discussion about this.
While the identification of the ants through the recovered reads matching the COI is a useful bonus, it is not definitive. The number of reads is small, and the VANA tool is not designed to recover non-encapsidated viruses. Additionally, if this experiment was to be repeated, it would be beneficial to have some morphological identification and/or a proper DNA barcoding on the ants (which would require collecting two samples for each species, one for the metagenome and one for the taxonomy).
Validity of the findings
It is clear that besides the Parvoviridae and Circoviridae families, the contigs extracted were mostly short and from different genomic regions. In some cases, this allowed for a taxonomic assignment, and presumably, in other cases, the contigs could only be used to make the Figure 1 (137 sequences were deposited on GenBank for the phylogenetic analyses out of the 22,406 contigs). But that is the nature of these metagenome studies. Therefore, I understand that the phylogeny used is there to illustrate that the virus contigs (or virus-like in some cases) fit into available taxonomies but it would be good to explain why neighbor joining method was chosen.
Additional comments
There are a few additional small edits:
Page 3 L23: The sentence needs to be rewritten as it reads as if the viruses have medical or agricultural relevance to human (instead of the host).
Page 4 L14: Densely forested tropical regions do not represent major interfaces but rather provides interface as a consequence of human activities surrounding these forests. Additionally, densely forested tropical regions clustered together, represent fewer interfaces than if the forests were scattered across a larger territory.
Page 4 L21: random/unbiased : all the tools relying on one animal will have preferred patterned, but those will be different to the human one. I like the way it is defined earlier “a less human-centric assessment of viral diversity at the ecosystem-scale”
Page 25 L4: Since endogenous paroviral elements are detected within invertebrate genomes, how many of the parvovirus contigs could be EPVs?
Figures with phylogenetic analyses (mostly 2 and 3): could you when the aligment is made on the protein or the nucleic acid and the size of the region aligned.
https://doi.org/10.24072/pci.infections.100077.rev12
