
Latest recommendations

| Id | Title * | Authors * ▲ | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
25 Apr 2023

The distribution, phenology, host range and pathogen prevalence of Ixodes ricinus in France: a systematic map and narrative reviewAn extensive review of Ixodes ricinus in European FranceRecommended by Ana Sofia Santos based on reviews by Ana Palomar and 1 anonymous reviewerTicks are obligate, bloodsucking, nonpermanent ectoparasitic arthropods. Among them, Ixodes ricinus is a classic example of an extreme generalist tick, presenting a highly permissive feeding behavior using different groups of vertebrates as hosts, such as mammalian (including humans), avian and reptilian species (Hoogstraal & Aeschlimann, 1982; Dantas-Torresa & Otranto, 2013). This ecological adaptation can account for the broad geographical distribution of I. ricinus populations, which extends from the western end of the European continent to the Ural Mountains in Russia, and from northern Norway to the Mediterranean basin, including the North African countries - Morocco, Algeria and Tunisia (https://ecdc.europa.eu/en/disease-vectors/surveillance-and-disease-data/tick-maps). The contact with different hosts also promotes the exposure/acquisition and transmission of various pathogenic agents (viruses, bacteriae, protists and nematodes) of veterinary and medical relevance (Aeschlimann et al., 1979). As one of the prime ticks found on humans, this species is implicated in diseases such as Lyme borreliosis, Spotted Fever Group rickettsiosis, Human Anaplasmosis, Human Babesiosis and Tick-borne Encephalitis (Velez et al., 2023). The climate change projections drawn for I. ricinus, in the scenario of global warming, point for the expansion/increase activity in both latitude and altitude (Medlock et al., 2013). The adequacy of vector modeling is relaying in the proper characterization of complex biological systems. Thus, it is essential to increase knowledge on I. ricinus, focusing on aspects such as genetic background, ecology and eco-epidemiology on a microscale but also at a country and region level, due to possible local adaptations of tick populations and genetic drift. In the present systematic revision, Perez et al. (2023) combine old and recently published data (mostly up to 2020) regarding I. ricinus distribution, phenology, host range and pathogen association in continental France and Corsica Island. Based on a keyword search of peer-reviewed papers on seven databases, as well as other sources of grey literature (mostly, thesis), the authors have synthesized information on: 1) Host parasitism to detect potential differences in host use comparing to other areas in Europe; 2) The spatiotemporal distribution of I. ricinus, to identify possible geographic trends in tick density, variation in activity patterns and the influence of environmental factors; 3) Tick-borne pathogens detected in this species, to better assess their spatial distribution and variation in exposure risk. As pointed out by both reviewers, this work clearly summarizes the information regarding I. ricinus and associated microorganisms from European France. This review also identifies remaining knowledge gaps, providing a comparable basis to orient future research. This is why I chose to recommend Perez et al (2023)'s preprint for Peer Community Infections. REFERENCES Aeschlimann, A., Burgdorfer, W., Matile, H., Peter, O., Wyler, R. (1979) Aspects nouveaux du rôle de vecteur joué par Ixodes ricinus L. en Suisse. Acta Tropica, 36, 181-191. Dantas-Torresa, F., Otranto, D. (2013) Seasonal dynamics of Ixodes ricinus on ground level and higher vegetation in a preserved wooded area in southern Europe. Veterinary Parasitology, 192, 253- 258. Hoogstraal, H., Aeschlimann, A. (1982) Tick-host specificity. Mitteilungen der Schweizerischen Entomologischen Gesellschaft, 55, 5-32. Medlock, J.M., Hansford, K.M., Bormane, A., Derdakova, M., Estrada-Peña, A., George, J.C., Golovljova, I., Jaenson, T.G.T., Jensen, J.K., Jensen, P.M., Kazimirova, M., Oteo, J.A., Papa, A., Pfister, K., Plantard, O., Randolph, S.E., Rizzoli, A., Santos-Silva, M.M., Sprong, H., Vial, L., Hendrickx, G., Zeller, H., Van Bortel, W. (2013) Driving forces for changes in geographical distribution of Ixodes ricinus ticks in Europe. Parasites and Vectors, 6. https://doi.org/10.1186/1756-3305-6-1 Perez, G., Bournez, L., Boulanger, N., Fite, J., Livoreil, B., McCoy, K., Quillery, E., René-Martellet, M., Bonnet, S. (2023) The distribution, phenology, host range and pathogen prevalence of Ixodes ricinus in France: a systematic map and narrative review. bioRxiv, ver. 1 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2023.04.18.537315 Velez, R., De Meeûs, T., Beati, L., Younsi, H., Zhioua, E., Antunes, S., Domingos, A., Ataíde Sampaio, D., Carpinteiro, D., Moerbeck, L., Estrada-Peña, A., Santos-Silva, M.M., Santos, A.S. (2023) Development and testing of microsatellite loci for the study of population genetics of Ixodes ricinus Linnaeus, 1758 and Ixodes inopinatus Estrada-Peña, Nava & Petney, 2014 (Acari: Ixodidae) in the western Mediterranean region. Acarologia, 63, 356-372. https://doi.org/10.24349/bvem-4h49 | The distribution, phenology, host range and pathogen prevalence of *Ixodes ricinus* in France: a systematic map and narrative review | Grégoire Perez, Laure Bournez, Nathalie Boulanger, Johanna Fite, Barbara Livoreil, Karen D. McCoy, Elsa Quillery, Magalie René-Martellet, and Sarah I. Bonnet | <p style="text-align: justify;">The tick <em>Ixodes ricinus</em> is the most important vector species of infectious diseases in European France. Understanding its distribution, phenology, and host species use, along with the distribution and preva... | | Animal diseases, Behaviour of hosts, infectious agents, or vectors, Disease Ecology/Evolution, Ecohealth, Ecology of hosts, infectious agents, or vectors, Epidemiology, Geography of infectious diseases, Interactions between hosts and infectious ag... | Ana Sofia Santos | 2022-12-06 14:52:44 | ||
28 May 2024
HIV self-testing positivity rate and linkage to confirmatory testing and care: a telephone survey in Côte d'Ivoire, Mali and SenegalThe benefits of HIV self-testing in West Africa: quantified.Recommended by Jessie Abbate based on reviews by 3 anonymous reviewersDespite decades of advances and understanding of the indiscriminate nature of human immunodeficiency virus (HIV), it remains shrouded in stigma that makes it difficult to reach some key populations at risk of transmission. The advent of self-testing technology for HIV (HIVST) has opened much-needed potential for bringing privacy to prevention that is crucial for curtailing its continued spread (Johnson et al., 2014). The HIV Self-Testing in Africa (STAR) Initiative (https://www.psi.org/fr/project/star/), carried out in Eastern and Southern Africa between 2015 and 2020 (Simwinga et al., 2022), demonstrated the market and public health operational potential of HIVST of different distribution methods. From 2019 to 2022, the “AutoTest de dépistage du VIH : Libre d’Accéder à la connaissance de son Statut" (ATLAS, translating to “HIVST: Freedom to know your status”) program built on these findings to quantify the public health value of HIVST for reaching key populations in West Africa (specifically, Mali, Senegal and Côte d’Ivoire) (Ky-Zerbo et al., 2022). The innovative secondary distribution methods these studies employed, where the primary targeted populations were also encouraged to take and provide tests to their contacts, helped widen the reach of HIVST within key population networks beyond those relying on access to HIV testing facilities.
The tricky part of the self-testing model lies in assessing its reach and impact while maintaining the privacy of self-testers that is central to its success. Following voluntary phone survey methods that previously were able to show expanded reach of HIVST to first-time testers in key populations in West Africa and high rates of confirmatory testing and treatment seeking (Kra et al., 2022), Kra et al. (Kra et al., 2024) quantified how many of these self-tests led to a positive result – allowing wider assessment of follow-up behaviors and positivity rates among the hard-to-reach populations the program had targeted.
While the numbers were low, the results were informative. Among respondents who reported a positive (“reactive”) HIVST, just 44% proceeded to confirmatory testing. This is lower than in other populations where HIVST follow-up has been assessed (Thirumurthy et al., 2016). The main reasons given for not confirming a reactive self-test was misinterpretation of HIVST results and not understanding that confirmatory testing was needed. The result thus highlighted a need for improved communication on how to correctly interpret HIVST results, and the authors provided ranges for how this misinterpretation could have affected their positivity estimates. However, the majority of those who sought confirmatory testing did so within 3 months, and nearly all of those with confirmed infection started on treatment. HIV positivity rates in the three countries were all higher than other published HIV positivity estimates (Giguère et al., 2021; Maheu-Giroux et al., 2019), suggesting that HIVST methods were highly effective at reaching the targeted communities. Finally, while the authors demonstrated their methods as an effective way of assessing the utility of HIVST campaigns and identifying ways to improve them, the follow-up surveys are likely too costly to replace current passive surveillance methods for assessing community disease burden. That said, these precious data should be taken as validation of the public health value of HIV self-testing in key populations across communities in West Africa. With improvements in communicating instructions for use and follow-up, there is little doubt that the innovation of HIVST primary and secondary distribution could become a widely useful addition to the fight against HIV.
References Giguère, K., Eaton, J. W., Marsh, K., Johnson, L. F., Johnson, C. C., Ehui, E., Jahn, A., Wanyeki, I., Mbofana, F., Bakiono, F., Mahy, M., & Maheu-Giroux, M. (2021). Trends in knowledge of HIV status and efficiency of HIV testing services in sub-Saharan Africa, 2000–20: a modelling study using survey and HIV testing programme data. The Lancet HIV, 8(5), e284–e293. https://doi.org/10.1016/S2352-3018(20)30315-5 Johnson, C., Baggaley, R., Forsythe, S., Van Rooyen, H., Ford, N., Napierala Mavedzenge, S., Corbett, E., Natarajan, P., & Taegtmeyer, M. (2014). Realizing the potential for HIV self-testing. In AIDS and Behavior (Vol. 18, Issue SUPPL. 4). Springer New York LLC. https://doi.org/10.1007/s10461-014-0832-x Kra, A. K., Fosto, A. S., N’guessan, K. N., Geoffroy, O., Younoussa, S., Kabemba, O. K., Gueye, P. A., Ndeye, P. D., Rouveau, N., Boily, M. C., Silhol, R., d’Elbée, M., Maheu-Giroux, M., Vautier, A., & Larmarange, J. (2022). Can HIV self-testing reach first-time testers? A telephone survey among self-test end users in Côte d’Ivoire, Mali, and Senegal. BMC Infectious Diseases, 22. https://doi.org/10.1186/s12879-023-08626-w Kra, A. K., Fotso, A. S., Rouveau, N., Maheu-Giroux, M., Boily, M.-C., Silhol, R., d’Elbée, M., Vautier, A., Lamarange, J., & the Atlas team. (2024). HIV self-testing positivity rate and linkage to confirmatory testing and care: a telephone survey in Côte d’Ivoire, Mali, and Senegal. MedRxiv, Ver. 4 Peer-Reviewed and Recommended by Peer Community in Infections, 2023.06.10.23291206. https://doi.org/https://doi.org/10.1101/2023.06.10.23291206 Ky-Zerbo, O., Desclaux, A., Boye, S., Maheu-Giroux, M., Rouveau, N., Vautier, A., Camara, C. S., Kouadio, B. A., Sow, S., Doumenc-Aidara, C., Gueye, P. A., Geoffroy, O., Kamemba, O. K., Ehui, E., Ndour, C. T., Keita, A., & Larmarange, J. (2022). “I take it and give it to my partners who will give it to their partners”: Secondary distribution of HIV self-tests by key populations in Côte d’Ivoire, Mali, and Senegal. BMC Infectious Diseases, 22. https://doi.org/10.1186/s12879-023-08319-4 Maheu-Giroux, M., Marsh, K., Doyle, C. M., Godin, A., Lanièce Delaunay, C., Johnson, L. F., Jahn, A., Abo, K., Mbofana, F., Boily, M. C., Buckeridge, D. L., Hankins, C. A., & Eaton, J. W. (2019). National HIV testing and diagnosis coverage in sub-Saharan Africa: A new modeling tool for estimating the “first 90” from program and survey data. AIDS, 33, S255–S269. https://doi.org/10.1097/QAD.0000000000002386 Simwinga, M., Gwanu, L., Hensen, B., Sigande, L., Mainga, M., Phiri, T., Mwanza, E., Kabumbu, M., Mulubwa, C., Mwenge, L., Bwalya, C., Kumwenda, M., Mubanga, E., Mee, P., Johnson, C. C., Corbett, E. L., Hatzold, K., Neuman, M., Ayles, H., & Taegtmeyer, M. (2022). Lessons learned from implementation of four HIV self-testing (HIVST) distribution models in Zambia: applying the Consolidated Framework for Implementation Research to understand impact of contextual factors on implementation. BMC Infectious Diseases, 22(Suppl 1). https://doi.org/10.1186/s12879-024-09168-5 Thirumurthy, H., Masters, S. H., Mavedzenge, S. N., Maman, S., Omanga, E., & Agot, K. (2016). Promoting male partner HIV testing and safer sexual decision making through secondary distribution of self-tests by HIV-negative female sex workers and women receiving antenatal and post-partum care in Kenya: a cohort study. The Lancet HIV, 3(6), e266–e274. https://doi.org/10.1016/S2352-3018(16)00041-2
| HIV self-testing positivity rate and linkage to confirmatory testing and care: a telephone survey in Côte d'Ivoire, Mali and Senegal | Kra Djuhe Arsene Kouassi, Arlette Simo Fotso, Nicolas Rouveau, Mathieu Maheu-Giroux, Marie-Claude Boily, Romain Silhol, Marc d'Elbee, Anthony Vautier, Joseph Larmarange, ATLAS Team | <p>HIV self-testing (HIVST) empowers individuals to decide when and where to test and with whom to share their results. From 2019 to 2022, the ATLAS program distributed ~ 400 000 HIVST kits in Côte d’Ivoire, Mali, and Senegal. It prioritised key p... | | Epidemiology | Jessie Abbate | 2023-06-16 16:40:51 | ||
07 Feb 2023

Three-way relationships between gut microbiota, helminth assemblages and bacterial infections in wild rodent populationsUnveiling the complex interactions between members of gut microbiomes: a significant advance provided by an exhaustive study of wild bank volesRecommended by Thomas Pollet based on reviews by Jason Anders and 1 anonymous reviewerThe gut of vertebrates is a host for hundreds or thousands of different species of microorganisms named the gut microbiome. This latter may differ greatly in natural environments between individuals, populations and species (1). The vertebrate gut microbiome plays key roles in host fitness through functions including nutrient acquisition, immunity and defense against infectious agents. While bank voles are small mammals potentially reservoirs of a large number of infectious agents, questions about the links between their gut microbiome and the presence of pathogens are scarcely addressed. In this study, Bouilloud et al. (2) used complementary analyses of community and microbial ecology to (i) assess the variability of gut bacteriome diversity and composition in wild populations of the bank vole Myodes glareolus collected in four different sites in Eastern France and (ii) evaluate the three-way interactions between the gut bacteriota, the gastro-intestinal helminths and pathogenic bacteria detected in the spleen. Authors identified important variations of the gut bacteriota composition and diversity among bank voles mainly explained by sampling localities. They found positive correlations between the specific richness of both the gut bacteria and the helminth community, as well as between the composition of these two communities, even when accounting for the influence of geographical distance. The helminths Aonchotheca murissylvatici, Heligmosomum mixtum and the bacteria Bartonella sp were the main taxa associated with the whole gut bacteria composition. Besides, changes in relative abundance of particular gut bacterial taxa were specifically associated with other helminths (Mastophorus muris, Catenotaenia henttoneni, Paranoplocephala omphalodes and Trichuris arvicolae) or pathogenic bacteria. Infections with Neoehrlichia mikurensis, Orientia sp, Rickettsia sp and P. omphalodes were especially associated with lower relative abundance of members of the family Erysipelotrichaceae (Firmicutes), while coinfections with higher number of bacterial infections were associated with lower relative abundance of members of the Bacteroidales family (Bacteroidetes). As pointed out by both reviewers, this study represents a significant advance in the field. I would like to commend the authors for this enormous work. The amount of data, analyses and results is considerable which has sometimes complicated the understanding of the story at the beginning of the evaluation process. Thanks to constructive scientific interactions with both reviewers through the two rounds of evaluation, the authors have efficiently addressed the reviewer's concerns and improved the manuscript, making this great story easier to read. The innovative results of this study emphasize the complex interlinkages between gut bacteriome and infections in wild animal populations and I strongly recommend this article for publication In Peer Community Infections. References (1) Vujkovic-Cvijin I, Sklar J, Jiang L, Natarajan L, Knight R, Belkaid Y (2020) Host variables confound gut microbiota studies of human disease. Nature, 587, 448–454. https://doi.org/10.1038/s41586-020-2881-9 (2) Bouilloud M, Galan M, Dubois A, Diagne C, Marianneau P, Roche B, Charbonnel N (2023) Three-way relationships between gut microbiota, helminth assemblages and bacterial infections in wild rodent populations. biorxiv, 2022.05.23.493084, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.05.23.493084 | Three-way relationships between gut microbiota, helminth assemblages and bacterial infections in wild rodent populations | Marie Bouilloud, Maxime Galan, Adelaide Dubois, Christophe Diagne, Philippe Marianneau, Benjamin Roche, Nathalie Charbonnel | <p>Background</p> <p>Despite its central role in host fitness, the gut microbiota may differ greatly between individuals. This variability is often mediated by environmental or host factors such as diet, genetics, and infections. Recently, a part... | | Disease Ecology/Evolution, Ecohealth, Interactions between hosts and infectious agents/vectors, Reservoirs, Zoonoses | Thomas Pollet | 2022-05-25 10:13:23 | ||
16 Jul 2024
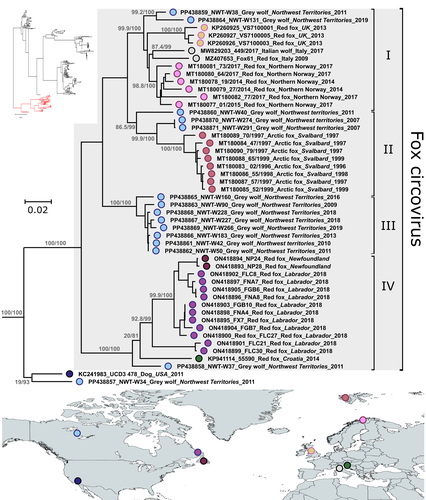
Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, CanadaWild canine viruses in the news. Better understanding multi-host transmission by adopting a disease ecology species community-based approachRecommended by Jean-Francois Guégan based on reviews by Arvind Varsani and 1 anonymous reviewerAccording to the international animal health authority, i.e., the World Organization on Animal Health (WOAH, former OIE), circoviruses are part of the Circoviridae family, which only includes 2 genera Circovirus and Cyclovirus, and infect swine, canine, ursid, viverrid, felid, pinniped, herpestid, mustelid, and several avian species (WOAH 2021). They are small (12–27 nm), non-enveloped, circular, single-stranded DNA viruses, viral replication is nuclear, and wild and domestic birds and mammals could serve as natural hosts. If most infections caused by circoviruses are subclinical in both wild and domestic species, they can be responsible for severe diseases in the commercial pig industry due to the Porcine circovirus-2 (PCV-2). These viruses can constitute a threat to wildlife, and cause their hosts to become immunocompromised, and animals often present with secondary coinfections. Canine circoviruses (CanineCV) harbour a worldwide distribution in dogs, and is the sole member of the viral genus to infect canines. They can be detected in wild carnivores, such as wolves, badgers, foxes and jackals, which indicates an ability for cross-species transmission between wildlife and domestic dogs. However, fox circovirus (FoCV), a distinct lineage of CanineCV, has been identified exclusively in wild canids (foxes and wolves) and not in dogs in Europe and North America, where it can cause in red foxes meningoencephalitis and other central nervous system signs. In their article, Canuti et al. (2024) investigate the presence, distribution and ecology of CanineCV in grey wolf specimens from the Northwest Territories, Canada. CanineCV occurrence appears to be relatively high with 45.3% positive specimens and parvoviral superinfections observed. The authors identify a high CanineCV genetic diversity among the investigated grey wolf specimens, and exacerbated by viral recombination. Phylogenetic analysis reveals the existence of 4 lineages, within each of them strains segregate by geography and not by host origin. This observed geographic segregation is interpreted as being due to the absence of exchange flows between grey wolf host subpopulations. Due to the paucity of knowledge on these circoviruses in wildlife and at the interface between wild and domestic animals, the authors discuss the plausible role of wolves as natural host reservoirs for disease transmission due to long-lasting virus-host coevolution. They are also conscious that additional maintenance hosts could exist in the wild, claiming for further studies to decipher fox circovirus disease ecology and transmission dynamics. This study underlines the importance of better understanding the transmission ecology and evolution of these Canine circoviruses, and I can only agree. Xiao et al. (2023), a research not referred to in the present work, evidenced CanineCV infection in cats in China, and obtained the first whole genome of cat-derived CanineCV. This emphasizes the importance of monitoring additional animal species and locations in the world to clarify disease ecology and transmission dynamics. A broader sampling of a wide range of animal species in different parts of the world using a species community-based approach is the key to understanding these CanineCV infections. References Marta CANUTI, Abigail V.L. KING, Giovanni FRANZO, H. Dean CLUFF, Lars E. LARSEN, Heather FENTON, Suzanne C. DUFOUR, Andrew S. LANG. 2024. Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, Canada. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2024.03.08.584028 World Organization on Animal Health. 2021. Circoviruses. https://www.woah.org/app/uploads/2021/05/circoviruses-infection-with.pdf [consulted on July 9th, 2024]. Xiangyu XIAO, Yan CHAO LI, Feng PEI XU, Xiangpi HAO, Shoujun LI, Pei ZHOU. 2023. Canine circovirus among dogs and cats in China: first identification in cats. Front. Microbiol. 14. https://doi.org/10.3389/fmicb.2023.1252272 | Diverse fox circovirus (*Circovirus canine*) variants circulate at high prevalence in grey wolves (*Canis lupus*) from the Northwest Territories, Canada | Marta Canuti, Abigail V.L. King, Giovanni Franzo, H. Dean Cluff, Lars E. Larsen, Heather Fenton, Suzanne C. Dufour, Andrew S. Lang | <p style="text-align: justify;">Canine circoviruses (CanineCV) have a worldwide distribution in dogs and are occasionally detected in wild carnivorans, indicating their ability for cross-species transmission. However, fox circovirus, a lineage of ... | | Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Epidemiology, Molecular genetics of hosts, infectious agents, or vectors, Population genetics of hosts, infectious agents, or vectors, Reservoirs, Taxonomy of hosts, infec... | Jean-Francois Guégan | Martine Peeters, Arvind Varsani | 2024-03-09 09:04:29 | |
27 Feb 2023
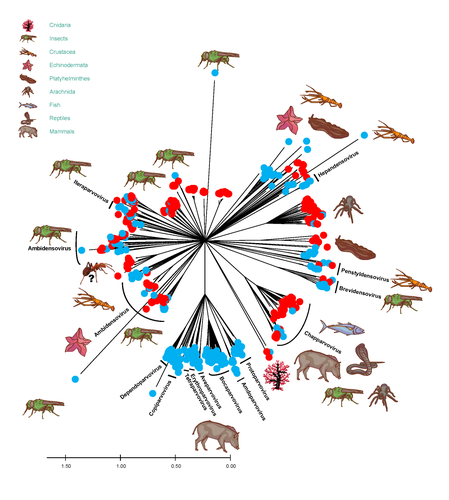
African army ants at the forefront of virome surveillance in a remote tropical forestA groundbreaking study using ants revealed a spectacular diversity of viruses in hardly accessible ecosystems like tropical forestsRecommended by Sebastien Massart based on reviews by Mart Krupovic and 1 anonymous reviewerDeciphering the virome (the set or assemblage of viruses) of the Earth, from individual organisms to entire ecosystems, has become a key priority. The first step to better understanding the impact of viruses on the ecology and functions of ecosystems is to describe their diversity. Such knowledge opens the gates to a better assessment of global nutrient cycling or of the threat that viruses represent to individual health. This explains the increasing number of pioneering studies that are currently sequencing the complete or partial genome of thousands of new viruses [1]. In their exciting study, Fritz and collaborators [2], authors sampled 209 army ants (Genus Dorylus) to investigate the virus diversity in dense forests that researchers cannot easily access. Indeed, these ants live in colonies (21 were sampled) that can move 1 km per day, covering a significant area and attacking many invertebrate and vertebrate preys. Each sample was sequenced by a protocol called VANA sequencing and allowing the enrichment of the sample in viral sequences [3], so improving the detection of viruses present at low abundance in the ant (and more specifically in its gut for viruses infecting preys). Around 45,000 contigs presented homologies with bacterial, plant, invertebrate, and vertebrate infecting viruses. Half could be assigned to 56 families and 157 genera of the International Committee on Taxonomy of Viruses. Beyond this amazing harvest of new and known virus sequences using an original methodology, the results significantly improve the current frontiers of known viral taxonomy and diversity and raise exciting research tracks to expand them. As a preprint, several blogs or news of leading scientists and journals have already highlighted this study. For example, in the news section of Science magazine, Jon Cohen underlined the originality of the approach for virus hunting on Earth with the title “Armed with air samplers, rope tricks, and—yes—ants, virus hunters spot threats in new ways”[4]. Another example is the mention of the publication by Elisabeth Bik in her Microbiome Digest: she wrote, “An amazing read is a fresh preprint from Fritz and collaborator describing an exciting method of sampling in difficult-to-reach environments“ [5]. The paper from Fritz et al [2] thus represents a significant advance in virus ecology, as already recognized by early readers, and this is why I strongly recommend its publication in PCI Infections. REFERENCES 1. Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, Lohr D, Novakovsky G, Buchfink B, Al-Shayeb B, Banfield JF, de la Peña M, Korobeynikov A, Chikhi R, Babaian A (2022) Petabase-scale sequence alignment catalyses viral discovery. Nature, 602, 142–147. https://doi.org/10.1038/s41586-021-04332-2 2. Fritz M, Reggiardo B, Filloux D, Claude L, Fernandez E, Mahé F, Kraberger S, Custer JM, Becquart P, Mebaley TN, Kombila LB, Lenguiya LH, Boundenga L, Mombo IM, Maganga GD, Niama FR, Koumba J-S, Ogliastro M, Yvon M, Martin DP, Blanc S, Varsani A, Leroy E, Roumagnac P (2023) African army ants at the forefront of virome surveillance in a remote tropical forest. bioRxiv, 2022.12.13.520061, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.12.13.520061 3. François S, Filloux D, Fernandez E, Ogliastro M, Roumagnac P (2018) Viral Metagenomics Approaches for High-Resolution Screening of Multiplexed Arthropod and Plant Viral Communities. In: Viral Metagenomics: Methods and Protocols Methods in Molecular Biology. (eds Pantaleo V, Chiumenti M), pp. 77–95. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7683-6_7 4. Cohen J (2023) Virus hunters test new surveillance tools. Science, 379, 16–17. https://doi.org/10.1126/science.adg5292 5. Ponsero A (2023) February 18th, 2023. Microbiome Digest - Bik’s Picks. https://microbiomedigest.com/2023/02/18/february-18th-2023/ | African army ants at the forefront of virome surveillance in a remote tropical forest | Matthieu Fritz, Berenice Reggiardo, Denis Filloux, Lisa Claude, Emmanuel Fernandez, Frederic Mahe, Simona Kraberger, Joy M. Custer, Pierre Becquart, Telstar Ndong Mebaley, Linda Bohou Kombila, Leadisaelle H. Lenguiya, Larson Boundenga, Illich M. M... | <p style="text-align: justify;">In this study, we used a predator-enabled metagenomics strategy to sample the virome of a remote and difficult-to-access densely forested African tropical region. Specifically, we focused our study on the use of arm... | | Ecohealth, Ecology of hosts, infectious agents, or vectors, One Health, Reservoirs, Viruses | Sebastien Massart | 2022-12-14 11:57:40 | ||
08 Aug 2023
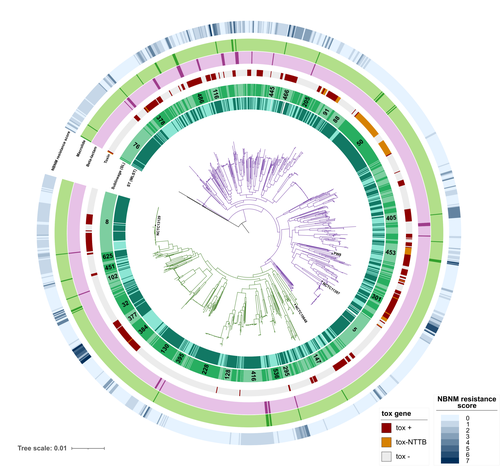
A global Corynebacterium diphtheriae genomic framework sheds light on current diphtheria reemergenceDIPHTOSCAN : A new tool for the genomic surveillance of diphtheriaRecommended by Rodolfo García-Contreras based on reviews by Ankur Mutreja and 2 anonymous reviewersOne of the greatest achievements of health sciences is the eradication of infectious diseases such as smallpox that in the past imposed a severe burden on humankind, through global vaccination campaigns. Moreover, progress towards the eradication of others such as poliomyelitis, dracunculiasis, and yaws is being made. In contrast, other infections that were previously contained are reemerging, due to several factors, including lack of access to vaccines due to geopolitical reasons, the rise of anti-vaccine movements, and the constant mobility of infected persons from the endemic sites. One of such disease is diphtheria, caused by Corynebacterium diphtheriae and a few other related species such as C. ulcerans and C. pseudotuberculosis. Importantly, in France, diphtheria cases reported in 2022 increased 7-fold from the average of previously recorded cases per year in the previous 4 years and the situation in other European countries is similar. Hence, as reported here, Hennart et al. (2023) developed DIPHTOSCAN, a free access bioinformatics tool with user-friendly interphase, aimed to easily identify, extract and interpret important genomic features such as the sublineage of the strain, the presence of the tox gene (as a string predictor for toxigenic disease) as well as genes coding other virulence factors such as fimbriae, and the presence of know resistant mechanisms towards antibiotics like penicillin and erythromycin currently used in the clinic to treat this infection. The authors validated the performance of their tool with a large collection of genomes, including those obtained from the isolates of the 2022 outbreak in France, more than 1,200 other genomes isolated from France, Algeria, and Yemen, and more than 500 genomes from several countries from Europe, America, Africa, Asia, and Oceania that are available through the NCBI site. DIPHTOSCAN will allow the rapid identification and surveillance of potentially dangerous strains such as those being tox-positive isolates and resistant to multiple drugs and/or first-line treatments and a better understanding of the epidemiology and evolution of this important reemerging disease. | A global *Corynebacterium diphtheriae* genomic framework sheds light on current diphtheria reemergence | Melanie Hennart, Chiara Crestani, Sebastien Bridel, Nathalie Armatys, Sylvie Brémont, Annick Carmi-Leroy, Annie Landier, Virginie Passet, Laure Fonteneau, Sophie Vaux, Julie Toubiana, Edgar Badell, Sylvain Brisse | <p style="text-align: justify;"><strong>Background</strong></p> <p style="text-align: justify;">Diphtheria, caused by <em>Corynebacterium diphtheriae</em>, reemerges in Europe since 2022. Genomic sequencing can inform on transmission routes and g... | | Drug resistance, tolerance and persistence, Epidemiology, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Microbiology of infections, Population genetics of hosts, infectiou... | Rodolfo García-Contreras | Ankur Mutreja | 2023-03-09 16:02:27 | |
23 Jan 2023

Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolenseWhole genome transcriptome reveals metabolic and immune susceptibility factors for Trypanosoma congolense infection in West-African livestockRecommended by Concepción Marañón based on reviews by 2 anonymous reviewersAfrican trypanosomiasis is caused by to the infection of a protozoan parasite of the Trypanosoma genus. It is transmitted by the tsetse fly, and is largely affecting cattle in the sub-humid areas of Africa, causing a high economic impact. However, not all the bovine strains are equally susceptible to the infection (1). In order to dissect the mechanisms underlying susceptibility to African trypanosoma infection, Peylhard et al (2) performed blood transcriptional profiles of trypanotolerant, trypanosensitive and mixed cattle breeds, before and after experimental infection with T. congolense. First of all, the authors have characterized the basal transcriptional profiles in the blood of the different breeds under study, which could be classified in a wide array of functional pathways. Of note, after infection some pathways were consistently enriched in all the group tested. Among them, the immune system-related ones were again on the top functions reported. The search for specific canonical pathways pointed to a prominent role of lipid and cholesterol-related pathways, as well as mitochondrial function and B and T lymphocyte activation. However, the analysis of infected animals demonstrated that trypanosusceptible animals showed a stronger transcriptomic reprogramming, highly enriched in specific metabolic and immunological pathways. It is worthy to highlight striking differences in genes involved in immune signal transduction, cytokines and markers of different leukocyte subpopulations. This work represents undoubtedly a significant momentum in the field, since the authors explore in deep a wide panel of cattle breeds representing the majority of West-African taurine and zebu in a systematic way. Since the animals were studied at different timepoints after infection, future longitudinal analyses of these datasets will be providing a precious insight on the kinetics of immune and metabolic reprogramming associated with susceptibility and tolerance to African trypanosoma infection, widening the application of this interesting study into new therapeutic interventions. References 1. Berthier D, Peylhard M, Dayo G-K, Flori L, Sylla S, Bolly S, Sakande H, Chantal I, Thevenon S (2015) A Comparison of Phenotypic Traits Related to Trypanotolerance in Five West African Cattle Breeds Highlights the Value of Shorthorn Taurine Breeds. PLOS ONE, 10, e0126498. https://doi.org/10.1371/journal.pone.0126498 2. Peylhard M, Berthier D, Dayo G-K, Chantal I, Sylla S, Nidelet S, Dubois E, Martin G, Sempéré G, Flori L, Thévenon S (2022) Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense. bioRxiv, 2022.06.10.495622, ver. 2 peer-reviewed and recommended by Peer Community Infections. https://doi.org/10.1101/2022.06.10.495622. | Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense | Moana Peylhard, David Berthier, Guiguigbaza-Kossigan Dayo, Isabelle Chantal, Souleymane Sylla, Sabine Nidelet, Emeric Dubois, Guillaume Martin, Guilhem Sempéré, Laurence Flori, Sophie Thévenon | <p>Animal African trypanosomosis, caused by blood protozoan parasites transmitted mainly by tsetse flies, represents a major constraint for millions of cattle in sub-Saharan Africa. Exposed cattle include trypanosusceptible indicine breeds, severe... | | Animal diseases, Genomics, functional genomics of hosts, infectious agents, or vectors, Resistance/Virulence/Tolerance | Concepción Marañón | Anonymous, Anonymous | 2022-06-14 17:06:57 | |
19 Feb 2024

Population genetics of Glossina palpalis gambiensis in the sleeping sickness focus of Boffa (Guinea) before and after eight years of vector control: no effect of control despite a significant decrease of human exposure to the diseaseReaching the last miles for transmission interruption of sleeping sickness in Guinea: follow-up of achievements and policy making using microsatellites-based population geneticsRecommended by Hugues Nana Djeunga based on reviews by Fabien HALKETT and 2 anonymous reviewers based on reviews by Fabien HALKETT and 2 anonymous reviewers
Thanks to the coordinated and sustained efforts of national control programs, the World Health Organization (WHO), bilateral cooperation and nongovernmental organizations, the incidence of Human African Trypanosomiasis (HAT), better known as sleeping sickness, has drastically decreased during the last two decades (WHO, 2023a). Indeed, between 1999 and 2022, the reported number of new cases of the chronic form of sleeping sickness (Trypanosoma brucei gambiense) fell by 97% (from 27 862 to 799), and the number of newly reported cases of the acute form of HAT (Trypanosoma brucei rhodesiense) fell by 94% (from 619 to 38) (WHO, 2023b). These encouraging trends led the WHO to target this debilitating and highly fatal (if untreated) vector-borne parasitic disease for elimination as a public health problem by 2020, and for interruption of transmission (zero case) by 2030 (WHO, 2021, WHO, 2023a). However, the disease is persisting in many foci, and even some cases of resurgence have been documented after unfortunate events such as war or pandemics (Moore et al., 1999; Sah et al., 2023. Simarro et al). Although effective control measures, diagnosis and treatment are complex and require specific skills (WHO, 2023), especially in a context which animal reservoirs, including hidden reservoirs, can contribute to the maintenance/persistence of infection (Welburn and Maudlin, 2012; Camara et al., 2021). Vector control therefore appears as a viable alternative to accelerate sleeping sickness transmission interruption, and WHO has identified some critical actions for HAT elimination, including the coordination of vector control and animal trypanosomiasis management among countries, stakeholders and other sectors (e.g. tourism and wildlife) through multisectoral national bodies to maximize synergies (WHO, 2021). The paper by Kagbadouno and Collaborators (2024) uses microsatellite markers genotyping and population genetics tools to investigate the impact of 11 years of tiny target-based vector control on the population biology of Glossina palpalis gambiensis in Boffa, one of the three active sleeping sickness foci in Guinea (Kagbadouno et al., 2012). Although vector control significantly reduced the apparent densities of tsetse flies (and therefore the human exposure to the vector) as well as the prevalence and incidence of the disease in the Boffa HAT focus (Courtin et al., 2015), no genetic signature of vector control was observed as no difference in population size, before and after the onset of the control policy, was found. The authors then provided national programs and implementing partners with indications on the actions to be taken to (i) maintain the achievements of vector control (thus avoiding rebound/resurgence as was experienced in the past (Franco et al., 2014), and (ii) accelerate the momentum towards elimination by for example combining these vector control efforts with medical surveys for case detection and treatment, in line with WHO recommendations (WHO, 2021). References Camara M, Soumah AM, Ilboudo H, Travaillé C, Clucas C, Cooper A, Kuispond Swar NR, Camara O, Sadissou I, Calvo Alvarez E, Crouzols A, Bart JM, Jamonneau V, Camara M, MacLeod A, Bucheton B, Rotureau B. Extravascular Dermal Trypanosomes in Suspected and Confirmed Cases of gambiense Human African Trypanosomiasis. Clin Infect Dis. 2021 Jul 1;73(1):12-20. https://doi.org/10.1093/cid/ciaa897 Courtin F, Camara M, Rayaisse JB, Kagbadouno M, Dama E, Camara O, Traore IS, Rouamba J, Peylhard M, Somda MB, Leno M, Lehane MJ, Torr SJ, Solano P, Jamonneau V, Bucheton B (2015) Reducing human-tsetse contact significantly enhances the efficacy of sleeping sickness active screening campaigns: a promising result in the context of elimination. PLoS Neglected Tropical Diseases, 9. https://doi.org/10.1371/journal.pntd.0003727 Franco JR, Simarro PP, Diarra A, Jannin JG. (2014) Epidemiology of human African trypanosomiasis. Clin Epidemiol. 6:257-75. https://doi.org/10.2147/CLEP.S39728 Kagbadouno, M. S., Séré, M., Ségard, A., Camara, A. D., Camara, M., Bucheton, B., ... & Ravel, S. (2023). Population genetics of Glossina palpalis gambiensis in the sleeping sickness focus of Boffa (Guinea) before and after eight years of vector control: no effect of control despite a significant decrease of human exposure to the disease. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2023.07.25.550445 Kagbadouno MS, Camara M, Rouamba J, Rayaisse JB, Traoré IS, Camara O, Onikoyamou MF, Courtin F, Ravel S, De Meeûs T, Bucheton B, Jamonneau V, Solano P (2012) Epidemiology of sleeping sickness in boffa (Guinea): where are the trypanosomes? PLoS Neglected Tropical Diseases, 6, e1949. https://doi.org/10.1371/journal.pntd.0001949 Moore A, Richer M, Enrile M, Losio E, Roberts J, Levy D. Resurgence of sleeping sickness in Tambura County, Sudan. Am J Trop Med Hyg. 1999 Aug;61(2):315-8. https://doi.org/10.4269/ajtmh.1999.61.315 Sah R, Mohanty A, Rohilla R, Padhi BK. A resurgence of Sleeping sickness amidst the COVID-19 pandemic: Correspondence. Int J Surg Open. 2023 Apr;53:100604. https://doi.org/10.1016/j.ijso.2023.100604 Welburn SC, Maudlin I. Priorities for the elimination of sleeping sickness. Adv Parasitol. 2012;79:299-337. https://doi.org/10.1016/B978-0-12-398457-9.00004-4 World Health Organization, 2021. Ending the neglect to attain the Sustainable Development Goals: a road map for neglected tropical diseases 2021–2030. World Health Organization, Geneva, Switzerland. ISBN: 978 92 4 001035 2. 196p. World Health Organization, 2023a. Trypanosomiasis, human African (sleeping sickness): key facts. Accessed at https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) on February 19, 2023. World Health Organization, 2023b. Human African Trypanosomiasis, (sleeping sickness): the global health observatory. Accessed at https://www.who.int/data/gho/data/themes/topics/human-african-trypanosomiasis on February 19, 2023. | Population genetics of *Glossina palpalis* gambiensis in the sleeping sickness focus of Boffa (Guinea) before and after eight years of vector control: no effect of control despite a significant decrease of human exposure to the disease | Moise S. Kagbadouno, Modou Séré, Adeline Ségard, Abdoulaye Dansy Camara, Mamadou Camara, Bruno Bucheton, Jean-Mathieu Bart, Fabrice Courtin, Thierry de Meeûs, Sophie Ravel | <p style="text-align: justify;">Human African trypanosomosis (HAT), also known as sleeping sickness, is still a major concern in endemic countries. Its cyclical vector are biting insects of the genus Glossina or tsetse flies. In Guinea, the mangro... | | Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Evolution of hosts, infectious agents, or vectors, Parasites, Population genetics of hosts, infectious agents, or vectors | Hugues Nana Djeunga | 2023-07-29 13:24:52 | ||
21 Jul 2022

Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV)Understanding the in vitro evolution of Cyprinid herpesvirus 3 (CyHV-3), a story of structural variations that can lead to the design of attenuated virus vaccinesRecommended by Jorge Amich based on reviews by Lucie Cappuccio and Veronique Hourdel
Structural variations (SVs) play a key role in viral evolution, and therefore they are also important for infection dynamics. However, the contribution of structural variations to the evolution of double-stranded viruses is limited. This knowledge can help to understand the population dynamics and might be crucial for the future development of viral attenuated vaccines. In this study, Fuandila et al (1) use the Cyprinid herpesvirus 3 (CyHV-3), commonly known as koi herpesvirus (KHV), to investigate the variability and contribution of structural variations (SV) for viral evolution after 99 passages in vitro. This virus, with the largest genome among herperviruses, causes a lethal infection in common carp and koi associated with mortalities up to 95% (2). Interestingly, KHV infections are caused by haplotype mixtures, which possibly are a source of genome diversification, but make genomic comparisons more difficult. The authors have used ultra-deep long-read sequencing of two passages, P78 and P99, which were previously described to have differences in virulence. They have found a surprisingly high and wide distribution of SVs along the genome, which were enriched in inversion and deletion events and that often led to defective viral genomes. Although it is known that these defective viral genomes negatively impact viral replication, their implications for virus persistence are still unclear. Subsequently, the authors concentrated on the virulence-relevant region ORF150, which was found to be different in P78 (deletion in 100% of the reads) and P99 (reference-like haplotype). To understand this loss and gain of full ORF150, they searched for SV turn-over in 10 intermediate passages. This analysis revealed that by passage 10 deleted and inverted (attenuated) haplotypes had already appeared, steadily increased frequency until P78, and then completely disappeared between P78 and P99. This is a striking result that raises new questions as to how this clearance occurs, which is really important as these reversions may result in undesirable increases in virulence of live-attenuated vaccines. We recommend this preprint because its use of ultra-deep long-read sequencing has permitted to better understand the role of SV diversity and dynamics in viral evolution. This study shows an unexpectedly high number of structural variations, revealing a novel source of virus diversification and confirming the different mixtures of haplotypes in different passages, including the gain of function. This research provides basic knowledge for the future design of live-attenuated vaccines, to prevent the reversion to virulent viruses. References (1) Fuandila NN, Gosselin-Grenet A-S, Tilak M-K, Bergmann SM, Escoubas J-M, Klafack S, Lusiastuti AM, Yuhana M, Fiston-Lavier A-S, Avarre J-C, Cherif E (2022) Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV). bioRxiv, 2022.03.10.483410, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.03.10.483410 (2) Sunarto A, McColl KA, Crane MStJ, Sumiati T, Hyatt AD, Barnes AC, Walker PJ. Isolation and characterization of koi herpesvirus (KHV) from Indonesia: identification of a new genetic lineage. Journal of Fish Diseases, 34, 87-101. https://doi.org/10.1111/j.1365-2761.2010.01216.x | Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV) | Nurul Novelia Fuandila, Anne-Sophie Gosselin-Grenet, Marie-Ka Tilak, Sven M Bergmann, Jean-Michel Escoubas, Sandro Klafack, Angela Mariana Lusiastuti, Munti Yuhana, Anna-Sophie Fiston-Lavier, Jean-Christophe Avarre, Emira Cherif | <p style="text-align: justify;">Structural variations (SVs) constitute a significant source of genetic variability in virus genomes. Yet knowledge about SV variability and contribution to the evolutionary process in large double-stranded (ds)DNA v... | | Animal diseases, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Viruses | Jorge Amich | Lucie Cappuccio, Veronique Hourdel | 2022-03-11 10:50:50 | |
24 Jan 2024
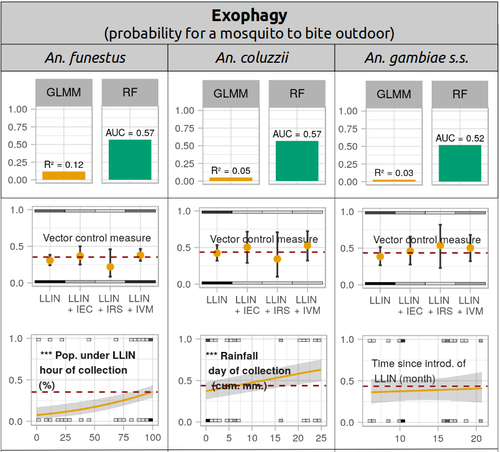
Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictabilityLarge and complete datasets, and modelling reveal the major determinants of physiological and behavioral insecticide resistance of malaria vectorsRecommended by Thierry DE MEEÛS based on reviews by Haoues Alout and 1 anonymous reviewer
Parasites represent the most diverse and adaptable ecological group of the biosphere (Timm & Clauson, 1988; De Meeûs et al., 1998; Poulin & Morand, 2000; De Meeûs & Renaud, 2002). The human species is known to considerably alter biodiversity, though it hosts, and thus sustains the maintenance of a spectacular diversity of parasites (179 species for eukaryotic species only) (De Meeûs et al., 2009). Among these, the five species of malaria agents (genus Plasmodium) remain a major public health issue around the world. Plasmodium falciparum is the most prevalent and lethal of these (Liu et al., 2010). With a pick of up to 2 million deaths due to malaria in 2004, deaths decreased to around 1 million in 2010 (Murray et al., 2012), to reach 619,000 in 2021, most of which in sub-Saharan Africa, and 79% of which were among children aged under 5 years (World Health Organization, 2022). As stressed by Taconet et al. (2023), reduction in malaria deaths is attributable to control measures, in particular against its vectors (mosquitoes of the genus Anopheles). Nevertheless, the success of vector control is hampered by several factors (biological, environmental and socio-economic), and in particular by the great propensity of targeted mosquitoes to evolve physiological or behavioral avoidance of anti-vectorial measures. In their paper Taconet et al. (2023) aims at understanding what are the main factors that determine the evolution of insecticide resistance in several malaria vectors, in relation to the biological determinisms of behavioral resistance and how fast such evolutions take place. To tackle these objectives, authors collected an impressive amount of data in two rural areas of West Africa. With appropriate modeling, Taconet et al. discovered, among many other results, a predominant role of public health measures, as compared to agricultural practices, in the evolution of physiological resistance. They also found that mosquito foraging activities are mostly explained by host availability and climate, with a poor, if any, association with genetic markers of physiological resistance to insecticides. These findings represent an important contribution to the field and should help at designing more efficient control strategies against malaria.
References De Meeûs T, Michalakis Y, Renaud F (1998) Santa Rosalia revisited: or why are there so many kinds of parasites in “the garden of earthly delights”? Parasitology Today, 14, 10–13. https://doi.org/10.1016/S0169-4758(97)01163-0 De Meeûs T, Prugnolle F, Agnew P (2009) Asexual reproduction in infectious diseases. In: Lost Sex: The Evolutionary Biology of Parthenogenesis (eds Schön I, Martens K, van Dijk P), pp. 517-533. Springer, NY. https://doi.org/10.1007/978-90-481-2770-2_24 De Meeûs T, Renaud F (2002) Parasites within the new phylogeny of eukaryotes. Trends in Parasitology, 18, 247–251. https://doi.org/10.1016/S1471-4922(02)02269-9 Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF, Ndjango JB, Sanz CM, Morgan DB, Locatelli S, Gonder MK, Kranzusch PJ, Walsh PD, Delaporte E, Mpoudi-Ngole E, Georgiev AV, Muller MN, Shaw GM, Peeters M, Sharp PM, Rayner JC, Hahn BH (2010) Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature, 467, 420–425. https://doi.org/10.1038/nature09442 Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. The Lancet, 379, 413–431. https://doi.org/10.1016/S0140-6736(12)60034-8 Poulin R, Morand S (2000) The diversity of parasites. Quarterly Review of Biology, 75, 277–293. https://doi.org/10.1086/393500 Taconet P, Soma DD, Zogo B, Mouline K, Simard F, Koffi AA, Dabire RK, Pennetier C, Moiroux N (2023) Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictability. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.20.504631 Timm RM, Clauson BL (1988) Coevolution: Mammalia. In: 1988 McGraw-Hill yearbook of science & technology, pp. 212–214. McGraw-Hill Book Company, New York. World Health Organization (2022) World malaria report 2022. Geneva: World Health Organization; 2022. Licence: CC BY-NC-SA 3.0 IGO. https://iris.who.int/bitstream/handle/10665/365169/9789240064898-eng.pdf?sequence=1.
| Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictability | Paul Taconet, Dieudonné Diloma Soma, Barnabas Zogo, Karine Mouline, Frédéric Simard, Alphonsine Amanan Koffi, Roch Kounbobr Dabiré, Cédric Pennetier, Nicolas Moiroux | <p>Insecticide resistance and behavioural adaptation of malaria mosquitoes affect the efficacy of long-lasting insecticide nets - currently the main tool for malaria vector control. To develop and deploy complementary, efficient and cost-effective... | | Behaviour of hosts, infectious agents, or vectors, Ecology of hosts, infectious agents, or vectors, Pesticide resistance, Population genetics of hosts, infectious agents, or vectors, Vectors | Thierry DE MEEÛS | Haoues Alout, Anonymous | 2023-07-03 11:29:10 |




