
Latest recommendations

| Id | Title▼ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
19 Jul 2023
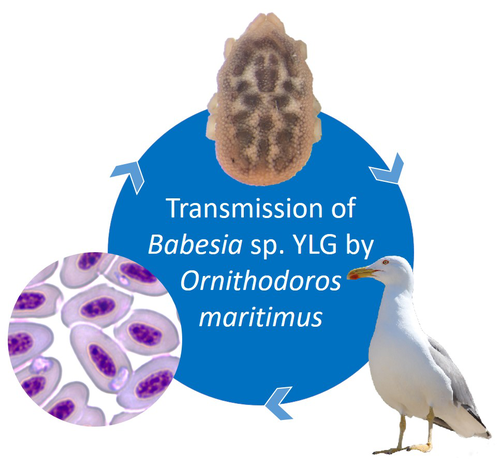
A soft tick vector of Babesia sp. YLG in Yellow-legged gull (Larus michahellis) nestsA four-year study reveals the potential role of the soft tick Ornithodoros maritimus in the transmission and circulation of Babesia sp. YLG in Yellow-legged gull colonies.Recommended by Thomas Pollet based on reviews by Hélène Jourdan and Tahar KernifWorldwide, ticks and tick-borne diseases are a persistent example of problems at the One Health interface between humans, wildlife, and environment (1, 2). The management and prevention of ticks and tick-borne diseases require a better understanding of host, tick and pathogen interactions and thus get a better view of the tick-borne pathosystems. In this study (3), the tick-borne pathosystem included three component species: first a seabird host, the Yellow-legged gull (YLG - Larus michahellis, Laridae), second a soft nidicolous tick (Ornithodoros maritimus, Argasidae, syn. Alectorobius maritimus) known to infest this host and third a blood parasite (Babesia sp. YLG, Piroplasmidae). In this pathosystem, authors investigated the role of the soft tick, Ornithodoros maritimus, as a potential vector of Babesia sp. YLG. They analyzed the transmission of Babesia sp. YLG by collecting different tick life stages from YLG nests during 4 consecutive years on the islet of Carteau (Gulf of Fos, Camargue, France). Ticks were dissected and organs were analyzed separately to detect the presence of Babesia sp DNA and to evaluate different transmission pathways. While the authors detected Babesia sp. YLG DNA in the salivary glands of nymphs, females and males, this result reveals a strong suspicion of transmission of the parasite by the soft tick. Babesia sp. YLG DNA was also found in tick ovaries, which could indicate possible transovarial transmission. Finally, the authors detected Babesia sp. YLG DNA in several male testes and in endospermatophores, and notably in a parasite-free female (uninfected ovaries and salivary glands). These last results raise the interesting possibility of sexual transmission from infected males to uninfected females. As pointed out by both reviewers, this is a nice study, well written and easy to read. All the results are new and allow to better understand the role of the soft tick, Ornithodoros maritimus, as a potential vector of Babesia sp. YLG. They finally question about the degree to which the parasite can be maintained locally by ticks and the epidemiological consequences of infection for both O. maritimus and its avian host. For all these reasons, I chose to recommend this article for Peer Community In Infections. References
| A soft tick vector of *Babesia* sp. YLG in Yellow-legged gull (*Larus michahellis*) nests | Claire Bonsergent, Marion Vittecoq, Carole Leray, Maggy Jouglin, Marie Buysse, Karen D. McCoy, Laurence Malandrin | <p style="text-align: justify;"><em>Babesia </em>sp. YLG has recently been described in Yellow-legged gull (<em>Larus michahellis</em>) chicks and belongs to the Peircei clade in the new classification of Piroplasms. Here, we studied <em>Babesia <... | | Ecology of hosts, infectious agents, or vectors, Eukaryotic pathogens/symbionts, Interactions between hosts and infectious agents/vectors, Parasites, Vectors | Thomas Pollet | 2023-03-29 14:33:40 | ||
08 Aug 2023
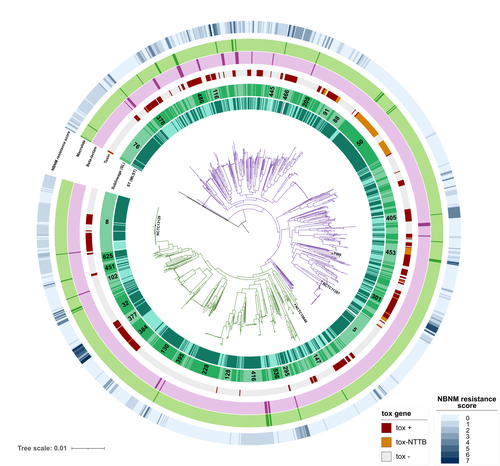
A global Corynebacterium diphtheriae genomic framework sheds light on current diphtheria reemergenceDIPHTOSCAN : A new tool for the genomic surveillance of diphtheriaRecommended by Rodolfo García-Contreras based on reviews by Ankur Mutreja and 2 anonymous reviewersOne of the greatest achievements of health sciences is the eradication of infectious diseases such as smallpox that in the past imposed a severe burden on humankind, through global vaccination campaigns. Moreover, progress towards the eradication of others such as poliomyelitis, dracunculiasis, and yaws is being made. In contrast, other infections that were previously contained are reemerging, due to several factors, including lack of access to vaccines due to geopolitical reasons, the rise of anti-vaccine movements, and the constant mobility of infected persons from the endemic sites. One of such disease is diphtheria, caused by Corynebacterium diphtheriae and a few other related species such as C. ulcerans and C. pseudotuberculosis. Importantly, in France, diphtheria cases reported in 2022 increased 7-fold from the average of previously recorded cases per year in the previous 4 years and the situation in other European countries is similar. Hence, as reported here, Hennart et al. (2023) developed DIPHTOSCAN, a free access bioinformatics tool with user-friendly interphase, aimed to easily identify, extract and interpret important genomic features such as the sublineage of the strain, the presence of the tox gene (as a string predictor for toxigenic disease) as well as genes coding other virulence factors such as fimbriae, and the presence of know resistant mechanisms towards antibiotics like penicillin and erythromycin currently used in the clinic to treat this infection. The authors validated the performance of their tool with a large collection of genomes, including those obtained from the isolates of the 2022 outbreak in France, more than 1,200 other genomes isolated from France, Algeria, and Yemen, and more than 500 genomes from several countries from Europe, America, Africa, Asia, and Oceania that are available through the NCBI site. DIPHTOSCAN will allow the rapid identification and surveillance of potentially dangerous strains such as those being tox-positive isolates and resistant to multiple drugs and/or first-line treatments and a better understanding of the epidemiology and evolution of this important reemerging disease. | A global *Corynebacterium diphtheriae* genomic framework sheds light on current diphtheria reemergence | Melanie Hennart, Chiara Crestani, Sebastien Bridel, Nathalie Armatys, Sylvie Brémont, Annick Carmi-Leroy, Annie Landier, Virginie Passet, Laure Fonteneau, Sophie Vaux, Julie Toubiana, Edgar Badell, Sylvain Brisse | <p style="text-align: justify;"><strong>Background</strong></p> <p style="text-align: justify;">Diphtheria, caused by <em>Corynebacterium diphtheriae</em>, reemerges in Europe since 2022. Genomic sequencing can inform on transmission routes and g... | | Drug resistance, tolerance and persistence, Epidemiology, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Microbiology of infections, Population genetics of hosts, infectiou... | Rodolfo García-Contreras | Ankur Mutreja | 2023-03-09 16:02:27 | |
27 Feb 2023
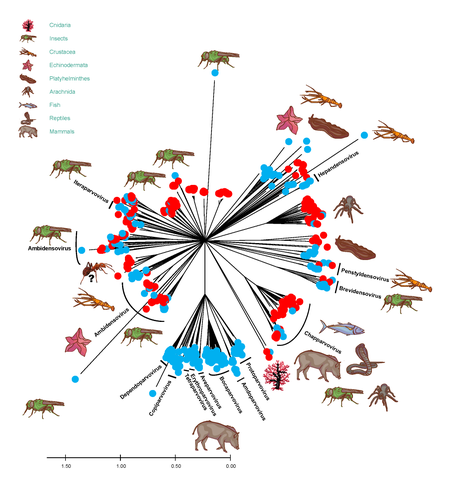
African army ants at the forefront of virome surveillance in a remote tropical forestA groundbreaking study using ants revealed a spectacular diversity of viruses in hardly accessible ecosystems like tropical forestsRecommended by Sebastien Massart based on reviews by Mart Krupovic and 1 anonymous reviewerDeciphering the virome (the set or assemblage of viruses) of the Earth, from individual organisms to entire ecosystems, has become a key priority. The first step to better understanding the impact of viruses on the ecology and functions of ecosystems is to describe their diversity. Such knowledge opens the gates to a better assessment of global nutrient cycling or of the threat that viruses represent to individual health. This explains the increasing number of pioneering studies that are currently sequencing the complete or partial genome of thousands of new viruses [1]. In their exciting study, Fritz and collaborators [2], authors sampled 209 army ants (Genus Dorylus) to investigate the virus diversity in dense forests that researchers cannot easily access. Indeed, these ants live in colonies (21 were sampled) that can move 1 km per day, covering a significant area and attacking many invertebrate and vertebrate preys. Each sample was sequenced by a protocol called VANA sequencing and allowing the enrichment of the sample in viral sequences [3], so improving the detection of viruses present at low abundance in the ant (and more specifically in its gut for viruses infecting preys). Around 45,000 contigs presented homologies with bacterial, plant, invertebrate, and vertebrate infecting viruses. Half could be assigned to 56 families and 157 genera of the International Committee on Taxonomy of Viruses. Beyond this amazing harvest of new and known virus sequences using an original methodology, the results significantly improve the current frontiers of known viral taxonomy and diversity and raise exciting research tracks to expand them. As a preprint, several blogs or news of leading scientists and journals have already highlighted this study. For example, in the news section of Science magazine, Jon Cohen underlined the originality of the approach for virus hunting on Earth with the title “Armed with air samplers, rope tricks, and—yes—ants, virus hunters spot threats in new ways”[4]. Another example is the mention of the publication by Elisabeth Bik in her Microbiome Digest: she wrote, “An amazing read is a fresh preprint from Fritz and collaborator describing an exciting method of sampling in difficult-to-reach environments“ [5]. The paper from Fritz et al [2] thus represents a significant advance in virus ecology, as already recognized by early readers, and this is why I strongly recommend its publication in PCI Infections. REFERENCES 1. Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, Lohr D, Novakovsky G, Buchfink B, Al-Shayeb B, Banfield JF, de la Peña M, Korobeynikov A, Chikhi R, Babaian A (2022) Petabase-scale sequence alignment catalyses viral discovery. Nature, 602, 142–147. https://doi.org/10.1038/s41586-021-04332-2 2. Fritz M, Reggiardo B, Filloux D, Claude L, Fernandez E, Mahé F, Kraberger S, Custer JM, Becquart P, Mebaley TN, Kombila LB, Lenguiya LH, Boundenga L, Mombo IM, Maganga GD, Niama FR, Koumba J-S, Ogliastro M, Yvon M, Martin DP, Blanc S, Varsani A, Leroy E, Roumagnac P (2023) African army ants at the forefront of virome surveillance in a remote tropical forest. bioRxiv, 2022.12.13.520061, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.12.13.520061 3. François S, Filloux D, Fernandez E, Ogliastro M, Roumagnac P (2018) Viral Metagenomics Approaches for High-Resolution Screening of Multiplexed Arthropod and Plant Viral Communities. In: Viral Metagenomics: Methods and Protocols Methods in Molecular Biology. (eds Pantaleo V, Chiumenti M), pp. 77–95. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7683-6_7 4. Cohen J (2023) Virus hunters test new surveillance tools. Science, 379, 16–17. https://doi.org/10.1126/science.adg5292 5. Ponsero A (2023) February 18th, 2023. Microbiome Digest - Bik’s Picks. https://microbiomedigest.com/2023/02/18/february-18th-2023/ | African army ants at the forefront of virome surveillance in a remote tropical forest | Matthieu Fritz, Berenice Reggiardo, Denis Filloux, Lisa Claude, Emmanuel Fernandez, Frederic Mahe, Simona Kraberger, Joy M. Custer, Pierre Becquart, Telstar Ndong Mebaley, Linda Bohou Kombila, Leadisaelle H. Lenguiya, Larson Boundenga, Illich M. M... | <p style="text-align: justify;">In this study, we used a predator-enabled metagenomics strategy to sample the virome of a remote and difficult-to-access densely forested African tropical region. Specifically, we focused our study on the use of arm... | | Ecohealth, Ecology of hosts, infectious agents, or vectors, One Health, Reservoirs, Viruses | Sebastien Massart | 2022-12-14 11:57:40 | ||
14 Feb 2024
A Bayesian analysis of birth pulse effects on the probability of detecting Ebola virus in fruit batsEpidemiological modeling to optimize the detection of zoonotic viruses in wild (reservoir) speciesRecommended by Aurelien Tellier based on reviews by Hetsron Legrace NYANDJO BAMEN and 1 anonymous reviewer based on reviews by Hetsron Legrace NYANDJO BAMEN and 1 anonymous reviewer
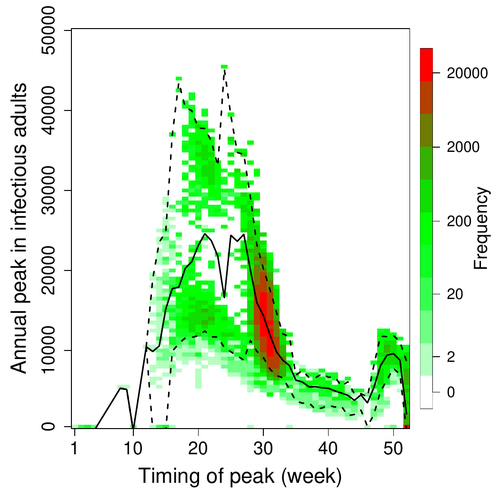
Various species of Ebolavirus have caused, and are still causing, zoonotic outbreaks and public health crises in Africa. Bats have long been hypothesized to be important reservoir populations for a series of viruses such as Hendra or Marburg viruses, the severe acute respiratory syndrome coronavirus (SARS-CoV, SARS-CoV-2) as well as Ebolaviruses [2, 3]. However the ecology of disease dynamics, disease transmission, and coevolution with their natural hosts of these viruses is still poorly understood, despite their importance for predicting novel outbreaks in human or livestock populations. The evidence that bats function as sylvatic reservoirs for Ebola viruses is yet only partial. Indeed, only few serological studies demonstrated the presence of Ebolavirus antibodies in young bats [4], albeit without providing positive controls of viral detection or identifying the viral species (via PCR). There is thus an unexplained discrepancy between serological data and viral detection [2, 4]. In this article, Pleydell et al. [1] use a modeling approach as well as published serological and age-structure (of the bat population) data to calibrate the model simulations. The study starts with the development of an age-structured epidemiological model which includes seasonal birth pulses and waning immunity, both generating pulses of Ebolavirus transmission within a colony of African straw-coloured fruit bats (Eidolon helvum). The epidemiological dynamics of such system of ordinary differential equations can generate annual outbreaks, skipped years or multi-annual cycles up to chaotic dynamics. Therefore, the calibration of the parameters, and the definition of biologically relevant priors, are key. To this aim, the serological data are obtained from a previous study in Cameroon [5], and the age structured of the bat population (birth and mortality) from a population study in Ghana [6]. These data are integrated into the Bayesian analysis and statistical framework to fit the model and generate predictions. In a nutshell, the authors show an overlap between the data and credibility intervals generated by the calibrated model, which thus explains well the seasonality of age-structure, namely changes in pup presence, number of lactating females, or proportion of juveniles in May. The authors can estimate that 76% of adults and 39% of young bats do survive each year, and infections are expected to last one and a half weeks. The epidemiological model predicts that annual birth pulses likely generate annual disease outbreaks, so that weeks 30 to 31 of each year, are predicted to be the best period to isolate the circulating Ebolavirus in this bat population. From the model predictions, the authors estimate the probability to have missed an infectious bat among all the samples tested by PCR being approximately of one per two thousands. The disease dynamics pattern observed in the serology data, and replicated by the model, is likely driven by seasonal pulses of young susceptible bats entering the population. This seasonal birth event increases the viral transmission, resulting in the observed peak of viral prevalence. With the inclusion of immunity waning and antibody persistence, the model results illuminate therefore why previous studies have detected only few positive cases by PCR tests, in contrast to the evidence from serological data. This study provides a first proof of principle that epidemiological modeling, despite its many simplifying assumptions, can be applied to wild species reservoirs of zoonotic diseases in order to optimize the design of field studies to detect viruses. Furthermore, such models can contribute to assess the probability and timing of zoonotic outbreaks in human or livestock populations. This article illustrates one of the manifold applications of mathematical theory of disease epidemiology to optimize sampling of pathogens/parasites or vaccine development and release [7, 8]. The further coupling of such models with population genetics theory and statistical inference methods (using parasite genome data) increasingly provide insights into the adaptation and evolution of parasites to human, crops and livestock populations [9, 10].
References [1] Pleydell D.R.J., Ndong Bass I., Mba Djondzo F.A., Djomsi D.M., Kouanfack C., Peeters M., and J. Cappelle. 2023. A Bayesian analysis of birth pulse effects on the probability of detecting Ebola virus in fruit bats. bioRxiv, ver. 3 peer reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2023.08.10.552777 [2] Caron A., Bourgarel M., Cappelle J., Liégeois F., De Nys H.M., and F. Roger. 2018. Ebola virus maintenance: if not (only) bats, what else? Viruses 10, 549. https://doi.org/10.3390/v10100549 [3] Letko M., Seifert S.N., Olival K.J., Plowright R.K., and V.J. Munster. 2020. Bat-borne virus diversity, spillover and emergence. Nature Reviews Microbiology 18, 461–471. https://doi.org/10.1038/s41579-020-0394-z [4] Leroy E.M., Kumulungui B., Pourrut X., Rouquet P., Hassanin A., Yaba P., Délicat A., Paweska J.T., Gonzalez J.P., and R. Swanepoel. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438, 575–576. https://doi.org/10.1038/438575a [5] Djomsi D.M. et al. 2022. Dynamics of antibodies to Ebolaviruses in an Eidolon helvum bat colony in Cameroon. Viruses 14, 560. https://doi.org/10.3390/v14030560 [6] Peel A.J. et al. 2016. Bat trait, genetic and pathogen data from large-scale investigations of African fruit bats Eidolon helvum. Scientific data 3, 1–11. https://doi.org/10.1038/sdata.2016.49 [7] Nyandjo Bamen H.L., Ntaganda J.M., Tellier A. and O. Menoukeu Pamen. 2023. Impact of imperfect vaccine, vaccine trade-off and population turnover on infectious disease dynamics. Mathematics, 11(5), p.1240. https://doi.org/10.3390/math11051240 [8] Saadi N., Chi Y.L., Ghosh S., Eggo R.M., McCarthy C.V., Quaife M., Dawa J., Jit M. and A. Vassall. 2021. Models of COVID-19 vaccine prioritisation: a systematic literature search and narrative review. BMC medicine, 19, pp.1-11. https://doi.org/10.1186/s12916-021-02190-3 [9] Maerkle, H., John S., Metzger, L., STOP-HCV Consortium, Ansari, M.A., Pedergnana, V. and Tellier, A., 2023. Inference of host-pathogen interaction matrices from genome-wide polymorphism data. bioRxiv, https://doi.org/10.1101/2023.07.06.547816. [10] Gandon S., Day T., Metcalf C.J.E. and B.T. Grenfell. 2016. Forecasting epidemiological and evolutionary dynamics of infectious diseases. Trends in ecology & evolution, 31(10), pp.776-788. https://doi.org/10.1016/j.tree.2016.07.010 | A Bayesian analysis of birth pulse effects on the probability of detecting Ebola virus in fruit bats | David R.J. Pleydell, Innocent Ndong Bass, Flaubert Auguste Mba Djondzo, Dowbiss Meta Djomsi, Charles Kouanfack, Martine Peeters, Julien Cappelle | <p>Since 1976 various species of Ebolavirus have caused a series of zoonotic outbreaks and public health crises in Africa. Bats have long been hypothesised to function as important hosts for ebolavirus maintenance, however the transmission ecology... | | Animal diseases, Disease Ecology/Evolution, Ecohealth, Ecology of hosts, infectious agents, or vectors, Epidemiology, Population dynamics of hosts, infectious agents, or vectors, Reservoirs, Viruses, Zoonoses | Aurelien Tellier | 2023-08-16 16:57:05 |




