
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers▲ | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
23 Jan 2023

Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolenseWhole genome transcriptome reveals metabolic and immune susceptibility factors for Trypanosoma congolense infection in West-African livestockRecommended by Concepción Marañón based on reviews by 2 anonymous reviewersAfrican trypanosomiasis is caused by to the infection of a protozoan parasite of the Trypanosoma genus. It is transmitted by the tsetse fly, and is largely affecting cattle in the sub-humid areas of Africa, causing a high economic impact. However, not all the bovine strains are equally susceptible to the infection (1). In order to dissect the mechanisms underlying susceptibility to African trypanosoma infection, Peylhard et al (2) performed blood transcriptional profiles of trypanotolerant, trypanosensitive and mixed cattle breeds, before and after experimental infection with T. congolense. First of all, the authors have characterized the basal transcriptional profiles in the blood of the different breeds under study, which could be classified in a wide array of functional pathways. Of note, after infection some pathways were consistently enriched in all the group tested. Among them, the immune system-related ones were again on the top functions reported. The search for specific canonical pathways pointed to a prominent role of lipid and cholesterol-related pathways, as well as mitochondrial function and B and T lymphocyte activation. However, the analysis of infected animals demonstrated that trypanosusceptible animals showed a stronger transcriptomic reprogramming, highly enriched in specific metabolic and immunological pathways. It is worthy to highlight striking differences in genes involved in immune signal transduction, cytokines and markers of different leukocyte subpopulations. This work represents undoubtedly a significant momentum in the field, since the authors explore in deep a wide panel of cattle breeds representing the majority of West-African taurine and zebu in a systematic way. Since the animals were studied at different timepoints after infection, future longitudinal analyses of these datasets will be providing a precious insight on the kinetics of immune and metabolic reprogramming associated with susceptibility and tolerance to African trypanosoma infection, widening the application of this interesting study into new therapeutic interventions. References 1. Berthier D, Peylhard M, Dayo G-K, Flori L, Sylla S, Bolly S, Sakande H, Chantal I, Thevenon S (2015) A Comparison of Phenotypic Traits Related to Trypanotolerance in Five West African Cattle Breeds Highlights the Value of Shorthorn Taurine Breeds. PLOS ONE, 10, e0126498. https://doi.org/10.1371/journal.pone.0126498 2. Peylhard M, Berthier D, Dayo G-K, Chantal I, Sylla S, Nidelet S, Dubois E, Martin G, Sempéré G, Flori L, Thévenon S (2022) Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense. bioRxiv, 2022.06.10.495622, ver. 2 peer-reviewed and recommended by Peer Community Infections. https://doi.org/10.1101/2022.06.10.495622. | Whole blood transcriptome profiles of trypanotolerant and trypanosusceptible cattle highlight a differential modulation of metabolism and immune response during infection by Trypanosoma congolense | Moana Peylhard, David Berthier, Guiguigbaza-Kossigan Dayo, Isabelle Chantal, Souleymane Sylla, Sabine Nidelet, Emeric Dubois, Guillaume Martin, Guilhem Sempéré, Laurence Flori, Sophie Thévenon | <p>Animal African trypanosomosis, caused by blood protozoan parasites transmitted mainly by tsetse flies, represents a major constraint for millions of cattle in sub-Saharan Africa. Exposed cattle include trypanosusceptible indicine breeds, severe... | | Animal diseases, Genomics, functional genomics of hosts, infectious agents, or vectors, Resistance/Virulence/Tolerance | Concepción Marañón | Anonymous, Anonymous | 2022-06-14 17:06:57 | |
29 Jan 2024
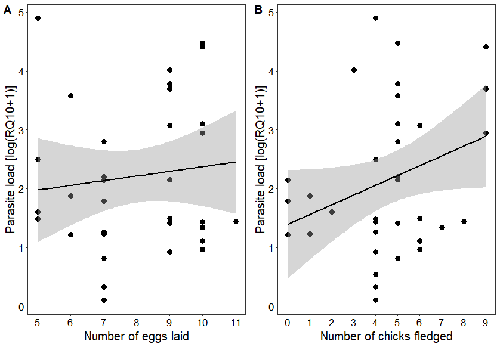
Spring reproductive success influences autumnal malarial load in a passerine birdAvian Plasmodium parasitaemia as an indicator of reproduction investmentRecommended by Claire Loiseau based on reviews by Luz García-Longoria and 2 anonymous reviewers based on reviews by Luz García-Longoria and 2 anonymous reviewers
Effects of the seasonal variations on within-host parasitaemia are still not well understood and potentially due to numerous factors, e.g. host and parasite species, host sex or age, or geographical regions. In this study, over three years in Switzerland, Pigeault et al. (2024) collected data on great tits reproductive outputs – laying date, clutch size, fledging success – to determine whether they were associated with avian Plasmodium parasitaemia before (winter), during (spring) and after (autumn) the breeding season. They focused on two lineages from two species: a highly generalist lineage Plasmodium relictum (lineage SGS1; Bensch et al. 2009) and a more specialized lineage Plasmodium homonucleophilum (lineage SW2). As previously found, they showed that parasitaemia level is low during the winter and then increase in spring (Applegate, 1970; Applegate 1971). Spring recurrences have been intensively studied but are still not well understood since many non-exclusive factors can provoke them, i.e environmental stressors, reproductive hormones, co-infections or bites of mosquitoes (Cornet et al. 2014). Interestingly, the parasitaemia level during the winter before and during the breeding season were not associated to the reproductive success, meaning that birds in their populations with low parasitaemia during the winter had not more fledglings than the ones with a higher parasitaemia. However, the individuals who invested the most in the reproduction with a higher number of fledglings had also a higher parasitaemia in the following autumn. The number of laid eggs was not associated with the parasitaemia during the following autumn, showing that the initial investment in the reproduction is less important than the parental care (e.g. chicks feeding) in terms of mid/long term cost. The originality here is that authors followed populations during three periods of the year, which is not an easy task and rarely done in natural populations. Their results highlight the mid/long-term effect of higher resource allocation into reproduction on individuals’ immune system and ability to control parasite replication. Further analyses on various lineages and bird populations from other geographical regions (i.e. different latitudes) would be the next relevant step. References Applegate JE (1971) Spring relapse of Plasmodium relictum infections in an experimental field population of English sparrows (Passer domesticus). Journal of Wildlife Diseases, 7, 37–42. https://doi.org/10.7589/0090-3558-7.1.37 Applegate JE, Beaudoin RL (1970) Mechanism of spring relapse in avian malaria: Effect of gonadotropin and corticosterone. Journal of Wildlife Diseases, 6, 443–447. https://doi.org/10.7589/0090-3558-6.4.443 Bensch S, Hellgren O, Pérez‐Tris J (2009) MalAvi: a public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Molecular Ecology Resources, 9, 1353-1358. https://doi.org/10.1111/j.1755-0998.2009.02692.x Cornet S, Nicot A, Rivero A, Gandon S (2014) Evolution of plastic transmission strategies in avian malaria. PLoS Pathogens, 10, e1004308. https://doi.org/10.1371/journal.ppat.1004308 Pigeault R, Cozzarolo CS, Wassef J, Gremion J, Bastardot M, Glaizot O, Christe P (2024) Spring reproductive success influences autumnal malarial load in a passerine bird. bioRxiv ver 3. Peer reviewed and recommended by Peer Community In Infections. https://doi.org/10.1101/2023.07.28.550923 | Spring reproductive success influences autumnal malarial load in a passerine bird | Romain Pigeault, Camille-Sophie Cozzarolo, Jérôme Wassef, Jérémy Gremion, Marc Bastardot, Olivier Glaizot, Philippe Christe | <p>Although avian haemosporidian parasites are widely used as model organisms to study fundamental questions in evolutionary and behavorial ecology of host-parasite interactions, some of their basic characteristics, such as seasonal variations in ... | | Interactions between hosts and infectious agents/vectors, Parasites | Claire Loiseau | Carolina Chagas, Anonymous, Luz García-Longoria | 2023-08-11 14:14:56 | |
06 Apr 2023

Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populationsChanges in aggressiveness in pathotypes of wheat leaf rustRecommended by Pierre Gladieux based on reviews by 2 anonymous reviewersUnderstanding the ecological and evolutionary factors underlying the spread of new fungal pathogen populations can inform the development of more effective management strategies. In plant pathology, pathogenicity is generally presented as having two components: ‘virulence’ (qualitative pathogenicity) and aggressiveness (quantitative pathogenicity). Changes in virulence in response to the deployment of new resistant varieties are a major driver of the spread of new populations (called pathotypes, or races) in modern agrosystems, and the genomic (i.e. proximal) and eco-evolutionary (i.e. ultimate) factors underlying these changes are well-documented [1,2,3]. By contrast, the role of changes in aggressiveness in the spread of pathotypes remains little known [4]. The study by Cécilia Fontyn and collaborators [5] set out to characterize changes in aggressiveness for isolates of two pathotypes of the wheat leaf rust (Puccinia triticina) that have been dominant in France during the 2005-2016 period. Isolates were genetically characterized using multilocus microsatellite typing and phenotypically characterized for three components of aggressiveness on wheat varieties: infection efficiency, latency period, and sporulation capacity. Using experiments that represent quite a remarkable amount of work and effort, Fontyn et al. showed that each dominant pathotype consisted of several genotypes, including common genotypes whose frequency changed over time. For each pathotype, the genotypes that were more common initially were replaced by a more aggressive genotype. Together, these results show that changes in the genetic composition of populations of fungal plant pathogens can be associated with, and may be caused by, changes in the quantitative components of pathogenicity. This study also illustrates how extensive, decade-long monitoring of fungal pathogen populations, such as the one conducted for wheat leaf rust in France, represents a very valuable resource for research. REFERENCES [1] Brown, J. K. (1994). Chance and selection in the evolution of barley mildew. Trends in Microbiology, 2(12), 470-475. https://doi.org/10.1016/0966-842x(94)90650-5 [2] Daverdin, G., Rouxel, T., Gout, L., Aubertot, J. N., Fudal, I., Meyer, M., Parlange, F., Carpezat, J., & Balesdent, M. H. (2012). Genome structure and reproductive behaviour influence the evolutionary potential of a fungal phytopathogen. PLoS Pathogens, 8(11), e1003020. https://doi.org/10.1371/journal.ppat.1003020 [3] Gladieux, P., Feurtey, A., Hood, M. E., Snirc, A., Clavel, J., Dutech, C., Roy, M., & Giraud, T. (2015). The population biology of fungal invasions.Molecular Ecology, 24(9), 1969-86. https://doi.org/10.1111/mec.13028 [4] Fontyn, C., Zippert, A. C., Delestre, G., Marcel, T. C., Suffert, F., & Goyeau, H. (2022). Is virulence phenotype evolution driven exclusively by Lr gene deployment in French Puccinia triticina populations?. Plant Pathology, 71(7), 1511-1524. https://doi.org/10.1111/ppa.13599 [5] Fontyn, C., Meyer, K. J., Boixel, A. L., Delestre, G., Piaget, E., Picard, C., Suffer, F., Marcel, T.C., & Goyeau, H. (2022). Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations. bioRxiv, 2022.08.29.505401, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.29.505401 | Evolution within a given virulence phenotype (pathotype) is driven by changes in aggressiveness: a case study of French wheat leaf rust populations | Cécilia FONTYN, Kevin JG MEYER, Anne-Lise BOIXEL, Ghislain DELESTRE, Emma PIAGET, Corentin PICARD, Frédéric SUFFERT, Thierry C MARCEL, Henriette GOYEAU | <p style="text-align: justify;">Plant pathogens are constantly evolving and adapting to their environment, including their host. Virulence alleles emerge, and then increase, and sometimes decrease in frequency within pathogen populations in respon... | | Coevolution, Epidemiology, Evolution of hosts, infectious agents, or vectors, Interactions between hosts and infectious agents/vectors, Pathogenic/Symbiotic Fungi, Phytopathology, Plant diseases, Population dynamics of hosts, infectious agents, or... | Pierre Gladieux | Emerson Del Ponte , Jacqui Shykoff, Leïla Bagny Beilhe , Alexey Mikaberidze | 2022-09-29 20:01:57 | |
24 Jan 2024
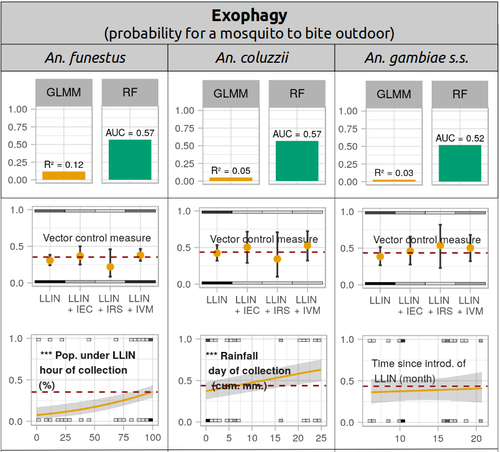
Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictabilityLarge and complete datasets, and modelling reveal the major determinants of physiological and behavioral insecticide resistance of malaria vectorsRecommended by Thierry DE MEEÛS based on reviews by Haoues Alout and 1 anonymous reviewer
Parasites represent the most diverse and adaptable ecological group of the biosphere (Timm & Clauson, 1988; De Meeûs et al., 1998; Poulin & Morand, 2000; De Meeûs & Renaud, 2002). The human species is known to considerably alter biodiversity, though it hosts, and thus sustains the maintenance of a spectacular diversity of parasites (179 species for eukaryotic species only) (De Meeûs et al., 2009). Among these, the five species of malaria agents (genus Plasmodium) remain a major public health issue around the world. Plasmodium falciparum is the most prevalent and lethal of these (Liu et al., 2010). With a pick of up to 2 million deaths due to malaria in 2004, deaths decreased to around 1 million in 2010 (Murray et al., 2012), to reach 619,000 in 2021, most of which in sub-Saharan Africa, and 79% of which were among children aged under 5 years (World Health Organization, 2022). As stressed by Taconet et al. (2023), reduction in malaria deaths is attributable to control measures, in particular against its vectors (mosquitoes of the genus Anopheles). Nevertheless, the success of vector control is hampered by several factors (biological, environmental and socio-economic), and in particular by the great propensity of targeted mosquitoes to evolve physiological or behavioral avoidance of anti-vectorial measures. In their paper Taconet et al. (2023) aims at understanding what are the main factors that determine the evolution of insecticide resistance in several malaria vectors, in relation to the biological determinisms of behavioral resistance and how fast such evolutions take place. To tackle these objectives, authors collected an impressive amount of data in two rural areas of West Africa. With appropriate modeling, Taconet et al. discovered, among many other results, a predominant role of public health measures, as compared to agricultural practices, in the evolution of physiological resistance. They also found that mosquito foraging activities are mostly explained by host availability and climate, with a poor, if any, association with genetic markers of physiological resistance to insecticides. These findings represent an important contribution to the field and should help at designing more efficient control strategies against malaria.
References De Meeûs T, Michalakis Y, Renaud F (1998) Santa Rosalia revisited: or why are there so many kinds of parasites in “the garden of earthly delights”? Parasitology Today, 14, 10–13. https://doi.org/10.1016/S0169-4758(97)01163-0 De Meeûs T, Prugnolle F, Agnew P (2009) Asexual reproduction in infectious diseases. In: Lost Sex: The Evolutionary Biology of Parthenogenesis (eds Schön I, Martens K, van Dijk P), pp. 517-533. Springer, NY. https://doi.org/10.1007/978-90-481-2770-2_24 De Meeûs T, Renaud F (2002) Parasites within the new phylogeny of eukaryotes. Trends in Parasitology, 18, 247–251. https://doi.org/10.1016/S1471-4922(02)02269-9 Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF, Ndjango JB, Sanz CM, Morgan DB, Locatelli S, Gonder MK, Kranzusch PJ, Walsh PD, Delaporte E, Mpoudi-Ngole E, Georgiev AV, Muller MN, Shaw GM, Peeters M, Sharp PM, Rayner JC, Hahn BH (2010) Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature, 467, 420–425. https://doi.org/10.1038/nature09442 Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. The Lancet, 379, 413–431. https://doi.org/10.1016/S0140-6736(12)60034-8 Poulin R, Morand S (2000) The diversity of parasites. Quarterly Review of Biology, 75, 277–293. https://doi.org/10.1086/393500 Taconet P, Soma DD, Zogo B, Mouline K, Simard F, Koffi AA, Dabire RK, Pennetier C, Moiroux N (2023) Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictability. bioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.08.20.504631 Timm RM, Clauson BL (1988) Coevolution: Mammalia. In: 1988 McGraw-Hill yearbook of science & technology, pp. 212–214. McGraw-Hill Book Company, New York. World Health Organization (2022) World malaria report 2022. Geneva: World Health Organization; 2022. Licence: CC BY-NC-SA 3.0 IGO. https://iris.who.int/bitstream/handle/10665/365169/9789240064898-eng.pdf?sequence=1.
| Physiological and behavioural resistance of malaria vectors in rural West-Africa : a data mining study to address their fine-scale spatiotemporal heterogeneity, drivers, and predictability | Paul Taconet, Dieudonné Diloma Soma, Barnabas Zogo, Karine Mouline, Frédéric Simard, Alphonsine Amanan Koffi, Roch Kounbobr Dabiré, Cédric Pennetier, Nicolas Moiroux | <p>Insecticide resistance and behavioural adaptation of malaria mosquitoes affect the efficacy of long-lasting insecticide nets - currently the main tool for malaria vector control. To develop and deploy complementary, efficient and cost-effective... | | Behaviour of hosts, infectious agents, or vectors, Ecology of hosts, infectious agents, or vectors, Pesticide resistance, Population genetics of hosts, infectious agents, or vectors, Vectors | Thierry DE MEEÛS | Haoues Alout, Anonymous | 2023-07-03 11:29:10 | |
21 Jul 2022

Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV)Understanding the in vitro evolution of Cyprinid herpesvirus 3 (CyHV-3), a story of structural variations that can lead to the design of attenuated virus vaccinesRecommended by Jorge Amich based on reviews by Lucie Cappuccio and Veronique Hourdel
Structural variations (SVs) play a key role in viral evolution, and therefore they are also important for infection dynamics. However, the contribution of structural variations to the evolution of double-stranded viruses is limited. This knowledge can help to understand the population dynamics and might be crucial for the future development of viral attenuated vaccines. In this study, Fuandila et al (1) use the Cyprinid herpesvirus 3 (CyHV-3), commonly known as koi herpesvirus (KHV), to investigate the variability and contribution of structural variations (SV) for viral evolution after 99 passages in vitro. This virus, with the largest genome among herperviruses, causes a lethal infection in common carp and koi associated with mortalities up to 95% (2). Interestingly, KHV infections are caused by haplotype mixtures, which possibly are a source of genome diversification, but make genomic comparisons more difficult. The authors have used ultra-deep long-read sequencing of two passages, P78 and P99, which were previously described to have differences in virulence. They have found a surprisingly high and wide distribution of SVs along the genome, which were enriched in inversion and deletion events and that often led to defective viral genomes. Although it is known that these defective viral genomes negatively impact viral replication, their implications for virus persistence are still unclear. Subsequently, the authors concentrated on the virulence-relevant region ORF150, which was found to be different in P78 (deletion in 100% of the reads) and P99 (reference-like haplotype). To understand this loss and gain of full ORF150, they searched for SV turn-over in 10 intermediate passages. This analysis revealed that by passage 10 deleted and inverted (attenuated) haplotypes had already appeared, steadily increased frequency until P78, and then completely disappeared between P78 and P99. This is a striking result that raises new questions as to how this clearance occurs, which is really important as these reversions may result in undesirable increases in virulence of live-attenuated vaccines. We recommend this preprint because its use of ultra-deep long-read sequencing has permitted to better understand the role of SV diversity and dynamics in viral evolution. This study shows an unexpectedly high number of structural variations, revealing a novel source of virus diversification and confirming the different mixtures of haplotypes in different passages, including the gain of function. This research provides basic knowledge for the future design of live-attenuated vaccines, to prevent the reversion to virulent viruses. References (1) Fuandila NN, Gosselin-Grenet A-S, Tilak M-K, Bergmann SM, Escoubas J-M, Klafack S, Lusiastuti AM, Yuhana M, Fiston-Lavier A-S, Avarre J-C, Cherif E (2022) Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV). bioRxiv, 2022.03.10.483410, ver. 4 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2022.03.10.483410 (2) Sunarto A, McColl KA, Crane MStJ, Sumiati T, Hyatt AD, Barnes AC, Walker PJ. Isolation and characterization of koi herpesvirus (KHV) from Indonesia: identification of a new genetic lineage. Journal of Fish Diseases, 34, 87-101. https://doi.org/10.1111/j.1365-2761.2010.01216.x | Structural variation turnovers and defective genomes: key drivers for the in vitro evolution of the large double-stranded DNA koi herpesvirus (KHV) | Nurul Novelia Fuandila, Anne-Sophie Gosselin-Grenet, Marie-Ka Tilak, Sven M Bergmann, Jean-Michel Escoubas, Sandro Klafack, Angela Mariana Lusiastuti, Munti Yuhana, Anna-Sophie Fiston-Lavier, Jean-Christophe Avarre, Emira Cherif | <p style="text-align: justify;">Structural variations (SVs) constitute a significant source of genetic variability in virus genomes. Yet knowledge about SV variability and contribution to the evolutionary process in large double-stranded (ds)DNA v... | | Animal diseases, Evolution of hosts, infectious agents, or vectors, Genomics, functional genomics of hosts, infectious agents, or vectors, Viruses | Jorge Amich | Lucie Cappuccio, Veronique Hourdel | 2022-03-11 10:50:50 | |
16 Jul 2024
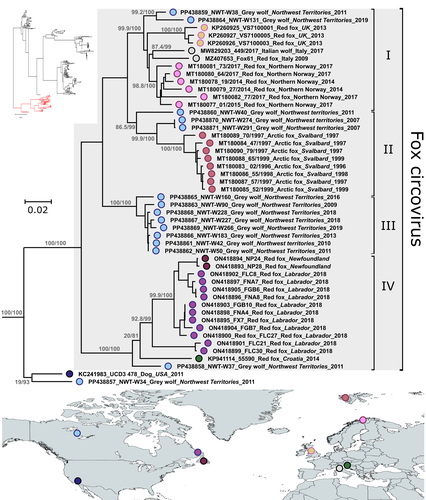
Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, CanadaWild canine viruses in the news. Better understanding multi-host transmission by adopting a disease ecology species community-based approachRecommended by Jean-Francois Guégan based on reviews by Arvind Varsani and 1 anonymous reviewerAccording to the international animal health authority, i.e., the World Organization on Animal Health (WOAH, former OIE), circoviruses are part of the Circoviridae family, which only includes 2 genera Circovirus and Cyclovirus, and infect swine, canine, ursid, viverrid, felid, pinniped, herpestid, mustelid, and several avian species (WOAH 2021). They are small (12–27 nm), non-enveloped, circular, single-stranded DNA viruses, viral replication is nuclear, and wild and domestic birds and mammals could serve as natural hosts. If most infections caused by circoviruses are subclinical in both wild and domestic species, they can be responsible for severe diseases in the commercial pig industry due to the Porcine circovirus-2 (PCV-2). These viruses can constitute a threat to wildlife, and cause their hosts to become immunocompromised, and animals often present with secondary coinfections. Canine circoviruses (CanineCV) harbour a worldwide distribution in dogs, and is the sole member of the viral genus to infect canines. They can be detected in wild carnivores, such as wolves, badgers, foxes and jackals, which indicates an ability for cross-species transmission between wildlife and domestic dogs. However, fox circovirus (FoCV), a distinct lineage of CanineCV, has been identified exclusively in wild canids (foxes and wolves) and not in dogs in Europe and North America, where it can cause in red foxes meningoencephalitis and other central nervous system signs. In their article, Canuti et al. (2024) investigate the presence, distribution and ecology of CanineCV in grey wolf specimens from the Northwest Territories, Canada. CanineCV occurrence appears to be relatively high with 45.3% positive specimens and parvoviral superinfections observed. The authors identify a high CanineCV genetic diversity among the investigated grey wolf specimens, and exacerbated by viral recombination. Phylogenetic analysis reveals the existence of 4 lineages, within each of them strains segregate by geography and not by host origin. This observed geographic segregation is interpreted as being due to the absence of exchange flows between grey wolf host subpopulations. Due to the paucity of knowledge on these circoviruses in wildlife and at the interface between wild and domestic animals, the authors discuss the plausible role of wolves as natural host reservoirs for disease transmission due to long-lasting virus-host coevolution. They are also conscious that additional maintenance hosts could exist in the wild, claiming for further studies to decipher fox circovirus disease ecology and transmission dynamics. This study underlines the importance of better understanding the transmission ecology and evolution of these Canine circoviruses, and I can only agree. Xiao et al. (2023), a research not referred to in the present work, evidenced CanineCV infection in cats in China, and obtained the first whole genome of cat-derived CanineCV. This emphasizes the importance of monitoring additional animal species and locations in the world to clarify disease ecology and transmission dynamics. A broader sampling of a wide range of animal species in different parts of the world using a species community-based approach is the key to understanding these CanineCV infections. References Marta CANUTI, Abigail V.L. KING, Giovanni FRANZO, H. Dean CLUFF, Lars E. LARSEN, Heather FENTON, Suzanne C. DUFOUR, Andrew S. LANG. 2024. Diverse fox circovirus (Circovirus canine) variants circulate at high prevalence in grey wolves (Canis lupus) from the Northwest Territories, Canada. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2024.03.08.584028 World Organization on Animal Health. 2021. Circoviruses. https://www.woah.org/app/uploads/2021/05/circoviruses-infection-with.pdf [consulted on July 9th, 2024]. Xiangyu XIAO, Yan CHAO LI, Feng PEI XU, Xiangpi HAO, Shoujun LI, Pei ZHOU. 2023. Canine circovirus among dogs and cats in China: first identification in cats. Front. Microbiol. 14. https://doi.org/10.3389/fmicb.2023.1252272 | Diverse fox circovirus (*Circovirus canine*) variants circulate at high prevalence in grey wolves (*Canis lupus*) from the Northwest Territories, Canada | Marta Canuti, Abigail V.L. King, Giovanni Franzo, H. Dean Cluff, Lars E. Larsen, Heather Fenton, Suzanne C. Dufour, Andrew S. Lang | <p style="text-align: justify;">Canine circoviruses (CanineCV) have a worldwide distribution in dogs and are occasionally detected in wild carnivorans, indicating their ability for cross-species transmission. However, fox circovirus, a lineage of ... | | Disease Ecology/Evolution, Ecology of hosts, infectious agents, or vectors, Epidemiology, Molecular genetics of hosts, infectious agents, or vectors, Population genetics of hosts, infectious agents, or vectors, Reservoirs, Taxonomy of hosts, infec... | Jean-Francois Guégan | Martine Peeters, Arvind Varsani | 2024-03-09 09:04:29 | |
14 Nov 2022
Ehrlichia ruminantium uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cellsAdhesion process of Ehrlichia ruminantium to its host cell: the role of the protein ERGACDS01230 elucidatedRecommended by Thomas Pollet based on reviews by Rodolfo García-Contreras and Alejandro Cabezas-CruzAs recently reported by the world organisation for animal health, 60% of infectious diseases are zoonotic with a significant part associated to ticks. Ticks can transmit various pathogens such as bacteria, viruses and parasites. Among pathogens known to be transmitted by ticks, Ehrlichia ruminantium is an obligate intracellular Gram-negative bacterium responsible for the fatal heartwater disease of domestic and wild ruminants (Allsopp, 2010). E. ruminantium is transmitted by ticks of the genus Amblyomma in the tropical and sub-Saharan areas, as well as in the Caribbean islands. It constitutes a major threat for the American livestock industries since a suitable tick vector is already present in the American mainland and potential introduction of infected A. variegatum through migratory birds or uncontrolled movement of animals from Caribbean could occur (i.e. Deem, 1998 ; Kasari et al 2010). The disease is also a major obstacle to the introduction of animals from heartwater-free to heartwater-infected areas into sub-Saharan Africa and thus restrains breeding programs aiming at upgrading local stocks (Allsopp, 2010). In this context, it is essential to develop control strategies against heartwater, as developing effective vaccines, for instance. Such an objective requires a better understanding of the early interaction of E. ruminantium and its host cells and of the mechanisms associated with bacterial adhesion to the host-cell. In this study, the authors. studied the role of E. ruminantium membrane protein ERGA_CDS_01230 in the adhesion process to host bovine aortic endothelial cells (BAEC). After successfully producing the recombinant version of the protein, Pinarello et al (2022) followed the in vitro culture of E. ruminantium in BAEC and observed that the expression of the protein peaked at the extracellular infectious elementary body stages. This result would suggest the likely involvement of the protein in the early interaction of E. ruminantium with its host cells. The authors then showed using flow cytometry, and scanning electron microscopy, that beads coated with the recombinant protein adhered to BAEC. In addition, they also observed that the adhesion protein of E. ruminantium interacted with proteins of the cell's lysate, membrane and organelle fractions. Additionally, enzymatic treatment, degrading dermatan and chondroitin sulfates on the surface of BAEC, was associated with a 50% reduction in the number of bacteria in the host cell after a development cycle, indicating that glycosaminoglycans might play a role in the adhesion of E. ruminantium to the host-cell. Finally, the authors observed that the adhesion protein of E. ruminantium induced a humoral response in vaccinated animals, making this protein a possible vaccine candidate. As rightly pointed out by both reviewers, the results of this study represent a significant advance (i) in the understanding of the role of the E. ruminantium membrane protein ERGA_CDS_01230 in the adhesion process to the host-cell and (ii) in the development of new control strategies against heartwater as this protein might potentially be used as an immunogen for the development of future vaccines. References Allsopp, B.A. (2010). Natural history of Ehrlichia ruminantium. Vet Parasitol 167, 123-135. https://doi.org/10.1016/j.vetpar.2009.09.014 Deem, S.L. (1998). A review of heartwater and the threat of introduction of Cowdria ruminantium and Amblyomma spp. ticks to the American mainland. J Zoo Wildl Med 29, 109-113. Kasari, T.R. et al (2010). Recognition of the threat of Ehrlichia ruminantium infection in domestic and wild ruminants in the continental United States. J Am Vet Med Assoc. 237:520-30. https://doi.org/10.2460/javma.237.5.520 Pinarello V, Bencurova E, Marcelino I, Gros O, Puech C, Bhide M, Vachiery N, Meyer DF (2022) Ehrlichia ruminantium uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cells. bioRxiv, 2021.06.15.447525, ver. 3 peer-reviewed and recommended by Peer Community in Infections. https://doi.org/10.1101/2021.06.15.447525 | *Ehrlichia ruminantium* uses its transmembrane protein Ape to adhere to host bovine aortic endothelial cells | Valérie Pinarello, Elena Bencurova, Isabel Marcelino, Olivier Gros, Carinne Puech, Mangesh Bhide, Nathalie Vachiery, Damien F. Meyer | <p><em>Ehrlichia ruminantium</em> is an obligate intracellular bacterium, transmitted by ticks of the genus <em>Amblyomma</em> and responsible for heartwater, a disease of domestic and wild ruminants. High genetic diversity of <em>E. ruminantium</... | | Interactions between hosts and infectious agents/vectors, Microbiology of infections | Thomas Pollet | Rodolfo García-Contreras, Alejandro Cabezas-Cruz | 2021-10-14 16:54:54 |




