
Recommendation
One of the greatest achievements of health sciences is the eradication of infectious diseases such as smallpox that in the past imposed a severe burden on humankind, through global vaccination campaigns. Moreover, progress towards the eradication of others such as poliomyelitis, dracunculiasis, and yaws is being made.
In contrast, other infections that were previously contained are reemerging, due to several factors, including lack of access to vaccines due to geopolitical reasons, the rise of anti-vaccine movements, and the constant mobility of infected persons from the endemic sites.
One of such disease is diphtheria, caused by Corynebacterium diphtheriae and a few other related species such as C. ulcerans and C. pseudotuberculosis. Importantly, in France, diphtheria cases reported in 2022 increased 7-fold from the average of previously recorded cases per year in the previous 4 years and the situation in other European countries is similar.
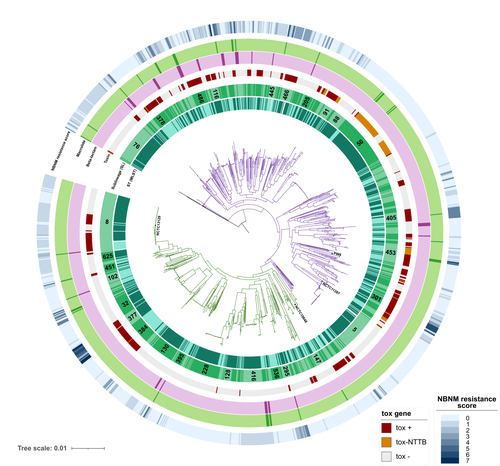
Hence, as reported here, Hennart et al. (2023) developed DIPHTOSCAN, a free access bioinformatics tool with user-friendly interphase, aimed to easily identify, extract and interpret important genomic features such as the sublineage of the strain, the presence of the tox gene (as a string predictor for toxigenic disease) as well as genes coding other virulence factors such as fimbriae, and the presence of know resistant mechanisms towards antibiotics like penicillin and erythromycin currently used in the clinic to treat this infection.
The authors validated the performance of their tool with a large collection of genomes, including those obtained from the isolates of the 2022 outbreak in France, more than 1,200 other genomes isolated from France, Algeria, and Yemen, and more than 500 genomes from several countries from Europe, America, Africa, Asia, and Oceania that are available through the NCBI site.
DIPHTOSCAN will allow the rapid identification and surveillance of potentially dangerous strains such as those being tox-positive isolates and resistant to multiple drugs and/or first-line treatments and a better understanding of the epidemiology and evolution of this important reemerging disease.
Reference
Hennart M., Crestani C., Bridel S., Armatys N., Brémont S., Carmi-Leroy A., Landier A., Passet V., Fonteneau L., Vaux S., Toubiana J., Badell E. and Brisse S. (2023). A global Corynebacterium diphtheriae genomic framework sheds light on current diphtheria reemergence. bioRxiv, 2023.02.20.529124, ver 3 peer-reviewed and recommended by PCI Infections. https://doi.org/10.1101/2023.02.20.529124
The authors have addressed my comments from the first round. I am happy to recommend the manuscript for publication.
https://doi.org/10.24072/pci.infections.100080.rev21DOI or URL of the preprint: https://doi.org/10.1101/2023.02.20.529124
Version of the preprint: 2
Dear authors, based on 2 independent reviews and in my own opinion your preprint is interesting and has merits, nevertheless, there are some issues you must fix in order to make it suitable for being recommended.
best regards,
Please go through my comments.
Summary:The authors of this study used DIPTHOSCAN to analyse the resurgence of diphtheria in France. The authors also added publically available datato develop a clearer picture. Using a combination of genome sequencing and phylogenetic analysis, they characterized the global population structure of the bacterium C. diphtheriae and identified factors that contribute to its ongoing spread and resurgence. The manuscript presents a valuable contribution to the field of infectious disease research, and the authors demonstrate a deep understanding of the complex genetic factors that underlie diphtheria's re-emergence.
Major comments:
1. Despite the author's assertion that DIPTHOSCAN is an easy-to-use tool, we observed certain issues in installing and using the publicly available DIPHTOSCAN tool that needs to be fixed. The fact that we found only the output file with information, such as species match, STs, 7 alleles, and tox types, led us to notice that some of the options listed were not executed.
2. Another major concern is that the manuscript focuses almost exclusively on genomic and genetic factors and does not give adequate attention to the epidemiological, vaccine escape and clinical aspects of diphtheria.
3. The authors in the manuscript should have emphasised the reason for the disease re-emergence and the role of pathogen-associated genes role in colonisation and disease transmission.
4. It would be valuable if authors could clarify the role of possible non-genetic factors in this resurgence of diphtheria, as well as evaluate the significance of their findings for public health policy and clinical practice.
Minor comments:
1. It is better to include the number of tox-negatives (line: 103).
2. While stating the number of sequenced strains in the aim (line: 105) and methods (line: 122-124; line: 128-130), the numbers do not match.
3. Typo error (line: 288) NTCT.
4. The authors may include more specific information on tox types and help readers comprehend how it differs from previous tox group classifications.
5. What is the fitness gained by some of the SL to be predominant for causing an increased number of infections?
6. While the authors present a wealth of data and analysis, for impact, the manuscript could be improved to make it a better read for a broader audience that doesn't understand the technicalities of phylogenomics.
Overall, this is an important and timely contribution to the field of infectious disease research, and the authors are to be commended for their comprehensive genomic analysis of diphtheria's re-emergence. With above comments addressed, I will support the acceptance of this work.
Bioinformatics tool and global population framework was used to analyzed C.diphtheriae genome. This work is very useful in examining the various genetic variants that frequently emerge in the C.diphtheriae infections. The sample size discussed was heterogenous and covered various sublineages. Overall, the study is an important contribution to the field.
https://doi.org/10.24072/pci.infections.100080.rev12The preprint by Hennart et al, presents an extensive work for the development of a new bioinformatics platform aimed to analyze C. diphtheriae genomes focusing in relevant data such as the presence of a functional tox gene, other genes encoding virulence factors, genes encoding antibiotic resistance and genes that allow biotype discrimination.
The platform performance was evaluated using different sets of genomes including several from recently isolated bacteria in a context of Diphtheria reemergence.
The manuscript is well written and well explained; however, my main criticism is that it is very long and too technical and hence will be difficult to understand by non-bioinformatics readers, I recommend to shorten the method section (perhaps send some material to the supplementary methods).
Also, although regarding the production of the Diphtheria toxin, the platform gave results that were mostly in agreement with the experimental determination of the gene presence by qPCR and its functionality (Elek test).
It predicted that only 50% of the non-toxigenic isolates were indeed non-toxigenic (L463), hence wrongly classifying 50% of the real non-toxigenic isolates as toxigenic.
And then in the discussion (L 659-661) you mention: “These cases may be attributable to (i) a lack of detection by the Elek test due to a low level of expression of the toxin gene in some strains, or (ii) yet unknown genetic mechanisms that abort tox gene expression entirely (unexplained true NTTB).”
Hence I would like to ask you what is the detection limit of Elek test? A very low expression of Tox would be equivalent to non-production?
Furthermore, perhaps these strains do not produce detectable toxin levels due defects in known tox gene regulation mechanisms, for example mutations in the regulatory region at the promoter level, or mutations in the main regulator gene encoding DtxR. Perhaps it mutates and becomes a supper repressor that represses the tox expression even in the absence of iron, etc.
Please add a section on tox gene regulation in the discussion and consider searching mutations in the regulatory regions of tox gene and in their regulators with your bioinformatics tools.
Minor:
1) L 162 “for 1 h DNA” add “,” after “h”.
2) Gene and species names should be in italic in the references.
https://doi.org/10.24072/pci.infections.100080.rev13